Proteins: how they provide striking evidence of design
https://reasonandscience.catsboard.com/t2062-proteins-how-they-provide-striking-evidence-of-design
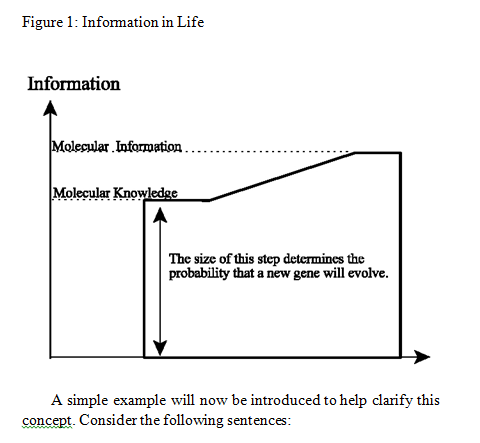
How ancestral proteins attained functionality, even though they were most likely markedly smaller than their contemporary descendants, remains a major, unresolved question in the origin of life. 10
Artur Gora (2013): On one hand, proteins need to have well-defined and organized structures in order to maintain stable functionality in the intracellular environment. On the other hand, some degree of flexibility is often required for catalytic activity. Molecular dynamics simulations have provided key insights into the importance of protein dynamics in catalysis, such as the observation of substrate access and product exit pathways that cannot be identified by inspecting crystal structures.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3744840/
Proteins are evidence of intelligent design par excellence. Instructional/specified complex information is required to get the right amino acid sequence which is essential to get functionality in vast sequence space ( amongst trillions os possible sequences, rare are the ones that provide function ), and every protein is irreducibly complex in the sense, that a minimal number of amino acids are required for each protein to get function. This constitutes a unsurmountable hurdle for the origin of life scenarios based on naturalistic hypotheses since unguided random events are too unspecific to get functional sequences in a viable timespan. Another true smack-down is the fact that single proteins or enzymes by themselves confer no advantage of survival at all, and have by their own no function. There is no reason why random RNA strands would become self-replicating. And even IF that were the case, so what? There would be no utility for them unless at least 50 different precisely arranged and correctly interlinked enzymes and proteins, each with its specific function, would interlink in a complex, just right metabolic network, and be encapsulated in a complex membrane with gates and pores, and in a precisely finely tuned and balanced homeostatic ambiance. Energy production and supply to each protein would also have to be fully set up right from the start...... hard to swallow. But if your wish of naturalism to be true is strong enough, just shut your reason up, and believe this. Blindly.
Proteins can be considered as information-enriched polymers. In fact, the instruction for the 3-D structure of a protein is embedded in the sequences of amino acids and the electrochemical attractive forces among them.
It is not surprising that various studies on evolving proteins have failed to show a viable mechanism. One study concluded that 10^63 attempts would be required to evolve a relatively short protein.
Functionally Acceptable Substitutions in Two a-Helical Regions of X Repressor
"The estimated number of sequences capable of adopting the h repressor fold is still 10^63 an exceedingly small fraction, about one in of the total number of possible 92-residue sequences."
http://sci-hub.tw/https://onlinelibrary.wiley.com/doi/pdf/10.1002/prot.340070403
And a similar result (10^65 attempts required) was obtained by comparing protein sequences. Another study found that 10^64 to 10^77 attempts are required, and another study concluded that 10^70 attempts would be required. So something like 10^70 attempts are required yet evolutionists estimate that only 10^43 attempts are possible. In other words, there is a shortfall of 27 orders of magnitude. But it gets worse. The estimate that 10^43 attempts are possible is utterly unrealistic. For it assumes billions of years are available, and that for that entire time the Earth is covered with bacteria, constantly churning out mutations and new protein experiments. Aside from the fact that these assumptions are entirely unrealistic, the estimate also suffers from the rather inconvenient fact that those bacteria are, err, full of proteins. In other words, for evolution to evolve proteins, they must already exist in the first place. This is absurd. And yet, even with these overly optimistic assumptions, evolution falls short by 27 orders of magnitude. The numbers don’t add up. Proteins reveal scientific problems for evolution. What is interesting is how evolutionists react to these problems.
https://darwins-god.blogspot.com.br/2017/04/new-book-new-proteins-evolve-very-easily.html
The protein that enables a firefly to glow, and also reproduce (as its illuminated abdomen also serves as a visible mating call), is a protein made up of a chain of 1,000 amino acids. The full range of possible proteins that can be coded with such a chain is 17 times the number of atoms in the visible universe. This number also represents the odds against the RANDOM coding of such a protein. Yet, DNA effortlessly assembles that protein, in the exactly correct, and absolutely necessary sequence and number of amino acids for the humble firefly. What are we to say of the 25,000 individual, highly specialized, absolutely necessary, and exactly correctly coded proteins in the human body? King David, perhaps, said it best: "We are fearfully and wonderfully made" (Psalms 139:14). "Time and Chance," as an explanation (read: cover story) for Life without a Creator, has all the scientific merit of the phrase, "Once Upon A Time."
A short protein molecule of 150 amino acids, the probability of building a 150 amino acids chain in which all linkages are peptide linkages would be roughly 1 chance in 10^45.
Paul Davies once said;
How did stupid atoms spontaneously write their own software … ? Nobody knows …… there is no known law of physics able to create information from nothing.
Dembsky : We also know from broad and repeated experience that intelligent agents can and do produce information-rich systems: we have positive experience-based knowledge of a cause that is sufficient to generate new instructing complex information, namely, intelligence. the design inference does not constitute an argument from ignorance. Instead, it constitutes an "inference to the best explanation" based upon our best available knowledge. It asserts the superior explanatory power of a proposed cause based upon its proven—its known—causal adequacy and based upon a lack of demonstrated efficacy among the competing proposed causes. The problem is that nature has too many options and without design couldn’t sort them all out. Natural mechanisms are too unspecific to determine any particular outcome. Mutation and natural selection or luck/chance/probability could theoretically form a new complex morphological feature like a leg or a limb with the right size and form , and arrange to find out the right body location to grow them, but it could also produce all kinds of other new body forms, and grow and attach them anywhere on the body, most of which have no biological advantage or are most probably deleterious to the organism. Natural mechanisms have no constraints, they could produce any kind of novelty. Its however that kind of freedom that makes it extremely unlikely that mere natural developments provide new specific evolutionary arrangements that are advantageous to the organism. Nature would have to arrange almost an infinite number of trials and errors until getting a new positive arrangement. Since that would become a highly unlikely event, design is a better explanation.
Even the simplest of these substances [proteins] represent extremely complex compounds, containing many thousands of atoms of carbon, hydrogen, oxygen, and nitrogen arranged in absolutely definite patterns, which are specific for each separate substance. To the student of protein structure the spontaneous formation of such an atomic arrangement in the protein molecule would seem as im- probable as would the accidental origin of the text of irgil’s “Aeneid” from scattered letter type.1
– A. I. Oparin
The argument of the proteins specified complexity
1. The number and sequence of amino acids in proteins, such as enzymes, are crucial.
2. Only specially-shaped forms (left-handed configurations) of each amino acid are used to form proteins.
3. Amino acids can be joined only by peptide bonds to form proteins.
4. To link together, each amino acid first must be activated by a specific enzyme.
5. Multiple special enzymes are required to bind messenger RNA to ribosomes before protein synthesis can begin or end.
6. Out of many details even these few have specified complexity without which the proteins could not exist. Not even half of the functional proteins could survive without important function.
7. If two amino acids are located within the peptidyl transferase center, they will easily form a peptide bond. But as soon as you do that in the absence of the ribosome, the ends of the amino acids come together, forming a cyclic structure. Polymers cannot form. But if the ends are kept apart, by a theoretical primitive ribosome, a chain of peptide bonds could grow into a polymer.
8. An irreducibly complex system cannot be produced gradually by slight, successive modifications of a precursor system, since any precursor to an irreducibly complex system is by definition nonfunctional. Since natural selection requires a function to select, an irreducibly complex biological system, if there is such a thing, would have to arise as an integrated unit for natural selection to have anything to act on. It is almost universally conceded that such a sudden event would be irreconcilable with the gradualism Darwin envisioned.
9. This is creation by an intelligent designer, and this is the dictionary meaning of the word God.
The individual macromolecules are complex
But the complex interaction of biological macromolecules is only one aspect of the problem facing the origin of life. What compounds the enigma is that the individual macromolecular components are themselves complex, in the sense that their sequences - of ribonucleotides in the case of RNA, or amino acids for proteins - are very specific. The linear amino acid sequence of a protein is specific because it must (a) be able to fold into a discrete 3-dimensional structure, and (b) have the right amino acids in the right positions in the linear sequence so that, when folded, they are in exactly the right positions in relation to each other to form the active site(s) of the protein. (And similar considerations apply to RNAs.) Sequences which meet these criteria are exceedingly rare compared with the astronomical number of possible sequences of a suitable length. For example Douglas Axe has estimated that only 1 in about 10^74 possible sequences will have biological function (Axe). So it is totally unrealistic to think that such sequences could have arisen by chance. How much less a suite of mutually dependent macromolecules? If the components themselves were not so improbable then it might be realistic to think that a complex combination of components could arise by chance; but the extreme improbability of the individual components is such that they are very unlikely to arise individually, and hence there is no chance whatever of an interdependent system. 5
Where even just two macromolecules are required to perform a function, then it would be necessary for both components to arise together: Because natural selection does not have foresight: if one component arises alone it will not be retained for potential future usefulness (when the second component is available), but will almost certainly degrade by mutation. And, it should be noted, if the probability of getting one component is 1 in 10^74 then the probability of getting two together is 1 in 10^148 (not 1 in 2x10^74); and so on for multi-component systems. This is why the obligatory mutual dependence of many macromolecules in even basic biological systems completely defies any hope of an evolutionary origin. So, in summary, the crux of the problem is that even a basic biological replicating system requires (a) several macromolecules with complementary functions with (b) each having a highly improbable sequence. And this combination of complexities presents an insurmountable challenge to a naturalistic origin of life.
Proteins need to interact together. They need interface compatibility. So this adds to the unsurmountable problem ot making them by chance. Lets say our good old friend chance , after 10^78 trial and errors , got a functional protein made. That function has imho no value, if it cannot interact properly with other proteins. Then our same old friend made another 10^78 trial and errors attempts, and got the second protein made. How many attempts would be required to make them compatible to be able to interact in a functional way ? So lets suppose for a moment, that they got the right interface compatibility, and interact properly . So what ? This interaction alone has a long way to go, to get all 20 protein complexes needed to get DNA replication done, and making them interact all together in a functional way. Its the same as to make a piston by trial and error. At the end you have eventually a device that is piston like. If you do not know imho exactly the size the piston has to have to fit into the motor block, nothing done. Nothing will go. So FORSIGHT and INTELLIGENCE in order to PROJECT the whole device is ESSENTIAL.
Dembsi, the design of life, general notes, page 11:
If the proportion of gene sequences that are biologically useful were large, there might be reason to think that point or chromosome mutations could be helpful in achieving the novel biological structures required by macroevolution. But all the evidence points to biologically useful gene sequences being exceedingly rare. It’s therefore highly unlikely that point and chromosome mutations can transform a duplicated gene into a novel functional gene. Genetic sequence space (i.e., the set of all possible genetic sequences) is functionally sparse (i.e., the overwhelming majority of genetic sequences don’t, and indeed can’t, do anything biologically useful or significant). As a consequence, navigating genetic sequence space by undirected means is no help getting from one island of functionality (i.e., one region of biologically useful or significant genetic sequences) to the next.
For instance, there is no evidence that conventional evolutionary mechanisms, such as natural selection, can evolve a gene in one region of genetic sequence space with one set of functions into a gene in a far distant region of genetic sequence space with another set of functions (distance here being measured in terms of sequence similarity). In the language of mathematical biology, genetic sequence space gives no indication of being highly interconnected by functional pathways that continuously connect genes with one function to genes with another (which would be required if natural selection, say, were to assist a duplicated gene in transforming into a novel gene). But there are still more problems with trying to make mutation the basis for macroevolution. For point and chromosome mutations to account for macroevolutionary change, it is not enough for individual genes to be transformed into novel genes that exhibit novel functions. Rather it is required that a whole suite of novel genes be produced through the coordinated transformation of existing genes. This is because for new biological structures to evolve (as required by macroevolution), many genes will have to change.
But it has not been demonstrated that mutations can produce the highly coordinated protein parts required for many biological structures. These are the structures that macroevolution would need to produce. Till now, however, there is no evidence for the coordinated macromutations required for macroevolution. The closest evidence cited in
textbooks includes increased immunity to malaria associated with the mutation for sickle-cell anemia and the resistance to antibiotics by mutant strains of bacteria.
In no such case, however, do we see a coordinated set of mutations that lead to complex novel structures. Sickle-cell anemia, for instance, is induced by a single point mutation that leads to single change in an amino acid (a valine is substituted for a glutamic acid in a hemoglobin molecule). Point mutations like this might enable organisms to stabilize and maintain themselves in the face of severe environmental pressure. In most instances, however, novel traits induced by such mutations do not continue to benefit the organisms when the environmental pressure is removed. Apart from environmental pressure, such mutations can even be deleterious. For instance, when an individual is heterozygous for sickle-cell anemia, the mutation provides an advantage for surviving the threat of malaria. It does so, however, at the expense of inflicting on homozygous individuals an anemia that impairs the transport of oxygen to the body’s cells. Indeed, sickle-cell anemia is often lethal.
To observe the origin of new species, one would expect to find it most readily in bacteria. That’s because bacteria can be easily mutated with chemicals and radiation in the laboratory, they take up very little space, and they have very short generation times. Indeed, thousands of mutations, billions of organisms, and thousands of generations can be studied by a single scientist. Yet, bacteriologists have never witnessed the origin of a new species. (Some new plant species have been observed to originate through hybridization, but the combining of two species to make a third is the opposite of the Darwinian process of splitting one species into two.) For mutations to contribute to evolution, they must benefit the organism. If a mutation harms the organism, it will tend to be eliminated, rather than favored, by natural selection. The only beneficial mutations that have ever been observed, in bacteria or in any other kind of organism, have been biochemical. That is, they affect only single molecules (such as the target molecule for streptomycin). There are no known beneficial mutations affecting morphology, or shape. All known morphological mutations are either neutral (i.e., they don’t have any noticeable effect on the organism’s fitness), or they are harmful—and the bigger their effect the more harmful they are. Yet, Darwinian evolution (i.e., the origin of new species, new organs, and new body plans) clearly requires changes in shape. So, there is no evidence for (and indeed a lot of evidence against) a role for mutations as providing raw materials for Darwinian evolution.
The specific genetic changes that give rise to the evolutionary origins of novel protein-protein interactions have rarely been documented in detail 6
Although numerous investigators assume that the global features of genetic networks are moulded by natural selection, there has been no formal demonstration of the adaptive origin of any genetic network 7 The mechanisms by which genetic networks become established evolutionarily are far from clear.
Many physicists, engineers and computer scientists, and some cell and developmental biologists, are convinced that biological networks exhibit properties that could only be products of natural selection; however, the matter has rarely been examined in the context of well-established evolutionary principles.
Alon states that it is “…wondrous that the solutions found by evolution have much in common with good engineering,”
Biological designs require a great deal more than the specification of a single DNA residue. Proteins, for instance, require a great many DNA residues to be specified. Even a single, typical protein requires more than 10^100 (a one followed by one hundred zeros) evolutionary experiments to find. This is an astronomical problem that is well beyond evolution’s capabilities. See here, here, here and here for more details. [url=10. http://darwins-god.blogspot.com.br/2011/06/vision-cascade-is-initiated-not-by.html]8[/url]
1) https://answersingenesis.org/origin-of-life/the-origin-of-life-dna-and-protein/
2) B.Alberts Molecular biology of the cell.
3) http://www.ncbi.nlm.nih.gov/pubmed/15321723
4) http://www.evolutionnews.org/2015/06/paper_reports_t096581.html
5) https://www.c4id.org.uk/index.php?option=com_content&view=article&id=211:the-problem-of-the-origin-of-life&catid=50:genetics&Itemid=43
6. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0001054
7. https://sci-hub.bz/http://www.nature.com/nrg/journal/v8/n10/abs/nrg2192.html
8. http://darwins-god.blogspot.com.br/2011/06/vision-cascade-is-initiated-not-by.html
9. Molecular Mechanisms of Autonomy in Biological Systems Relativity of Code, Energy and Mass, page 34
10. https://www.nasa.gov/sites/default/files/atoms/files/pohorille_wilson_shannon_2017_1.pdf
More readings:
Comprehensive experimental fitness landscape and evolutionary network for small RNA
Our study suggests that replaying the “tape of life” at the very origin of life might lead to quite different results.
https://reasonandscience.catsboard.com/t2062-proteins-how-they-provide-striking-evidence-of-design
How ancestral proteins attained functionality, even though they were most likely markedly smaller than their contemporary descendants, remains a major, unresolved question in the origin of life. 10
Artur Gora (2013): On one hand, proteins need to have well-defined and organized structures in order to maintain stable functionality in the intracellular environment. On the other hand, some degree of flexibility is often required for catalytic activity. Molecular dynamics simulations have provided key insights into the importance of protein dynamics in catalysis, such as the observation of substrate access and product exit pathways that cannot be identified by inspecting crystal structures.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3744840/
Proteins are evidence of intelligent design par excellence. Instructional/specified complex information is required to get the right amino acid sequence which is essential to get functionality in vast sequence space ( amongst trillions os possible sequences, rare are the ones that provide function ), and every protein is irreducibly complex in the sense, that a minimal number of amino acids are required for each protein to get function. This constitutes a unsurmountable hurdle for the origin of life scenarios based on naturalistic hypotheses since unguided random events are too unspecific to get functional sequences in a viable timespan. Another true smack-down is the fact that single proteins or enzymes by themselves confer no advantage of survival at all, and have by their own no function. There is no reason why random RNA strands would become self-replicating. And even IF that were the case, so what? There would be no utility for them unless at least 50 different precisely arranged and correctly interlinked enzymes and proteins, each with its specific function, would interlink in a complex, just right metabolic network, and be encapsulated in a complex membrane with gates and pores, and in a precisely finely tuned and balanced homeostatic ambiance. Energy production and supply to each protein would also have to be fully set up right from the start...... hard to swallow. But if your wish of naturalism to be true is strong enough, just shut your reason up, and believe this. Blindly.
Proteins can be considered as information-enriched polymers. In fact, the instruction for the 3-D structure of a protein is embedded in the sequences of amino acids and the electrochemical attractive forces among them.
It is not surprising that various studies on evolving proteins have failed to show a viable mechanism. One study concluded that 10^63 attempts would be required to evolve a relatively short protein.
Functionally Acceptable Substitutions in Two a-Helical Regions of X Repressor
"The estimated number of sequences capable of adopting the h repressor fold is still 10^63 an exceedingly small fraction, about one in of the total number of possible 92-residue sequences."
http://sci-hub.tw/https://onlinelibrary.wiley.com/doi/pdf/10.1002/prot.340070403
And a similar result (10^65 attempts required) was obtained by comparing protein sequences. Another study found that 10^64 to 10^77 attempts are required, and another study concluded that 10^70 attempts would be required. So something like 10^70 attempts are required yet evolutionists estimate that only 10^43 attempts are possible. In other words, there is a shortfall of 27 orders of magnitude. But it gets worse. The estimate that 10^43 attempts are possible is utterly unrealistic. For it assumes billions of years are available, and that for that entire time the Earth is covered with bacteria, constantly churning out mutations and new protein experiments. Aside from the fact that these assumptions are entirely unrealistic, the estimate also suffers from the rather inconvenient fact that those bacteria are, err, full of proteins. In other words, for evolution to evolve proteins, they must already exist in the first place. This is absurd. And yet, even with these overly optimistic assumptions, evolution falls short by 27 orders of magnitude. The numbers don’t add up. Proteins reveal scientific problems for evolution. What is interesting is how evolutionists react to these problems.
https://darwins-god.blogspot.com.br/2017/04/new-book-new-proteins-evolve-very-easily.html
The protein that enables a firefly to glow, and also reproduce (as its illuminated abdomen also serves as a visible mating call), is a protein made up of a chain of 1,000 amino acids. The full range of possible proteins that can be coded with such a chain is 17 times the number of atoms in the visible universe. This number also represents the odds against the RANDOM coding of such a protein. Yet, DNA effortlessly assembles that protein, in the exactly correct, and absolutely necessary sequence and number of amino acids for the humble firefly. What are we to say of the 25,000 individual, highly specialized, absolutely necessary, and exactly correctly coded proteins in the human body? King David, perhaps, said it best: "We are fearfully and wonderfully made" (Psalms 139:14). "Time and Chance," as an explanation (read: cover story) for Life without a Creator, has all the scientific merit of the phrase, "Once Upon A Time."
A short protein molecule of 150 amino acids, the probability of building a 150 amino acids chain in which all linkages are peptide linkages would be roughly 1 chance in 10^45.
Paul Davies once said;
How did stupid atoms spontaneously write their own software … ? Nobody knows …… there is no known law of physics able to create information from nothing.
Dembsky : We also know from broad and repeated experience that intelligent agents can and do produce information-rich systems: we have positive experience-based knowledge of a cause that is sufficient to generate new instructing complex information, namely, intelligence. the design inference does not constitute an argument from ignorance. Instead, it constitutes an "inference to the best explanation" based upon our best available knowledge. It asserts the superior explanatory power of a proposed cause based upon its proven—its known—causal adequacy and based upon a lack of demonstrated efficacy among the competing proposed causes. The problem is that nature has too many options and without design couldn’t sort them all out. Natural mechanisms are too unspecific to determine any particular outcome. Mutation and natural selection or luck/chance/probability could theoretically form a new complex morphological feature like a leg or a limb with the right size and form , and arrange to find out the right body location to grow them, but it could also produce all kinds of other new body forms, and grow and attach them anywhere on the body, most of which have no biological advantage or are most probably deleterious to the organism. Natural mechanisms have no constraints, they could produce any kind of novelty. Its however that kind of freedom that makes it extremely unlikely that mere natural developments provide new specific evolutionary arrangements that are advantageous to the organism. Nature would have to arrange almost an infinite number of trials and errors until getting a new positive arrangement. Since that would become a highly unlikely event, design is a better explanation.
Even the simplest of these substances [proteins] represent extremely complex compounds, containing many thousands of atoms of carbon, hydrogen, oxygen, and nitrogen arranged in absolutely definite patterns, which are specific for each separate substance. To the student of protein structure the spontaneous formation of such an atomic arrangement in the protein molecule would seem as im- probable as would the accidental origin of the text of irgil’s “Aeneid” from scattered letter type.1
– A. I. Oparin
The argument of the proteins specified complexity
1. The number and sequence of amino acids in proteins, such as enzymes, are crucial.
2. Only specially-shaped forms (left-handed configurations) of each amino acid are used to form proteins.
3. Amino acids can be joined only by peptide bonds to form proteins.
4. To link together, each amino acid first must be activated by a specific enzyme.
5. Multiple special enzymes are required to bind messenger RNA to ribosomes before protein synthesis can begin or end.
6. Out of many details even these few have specified complexity without which the proteins could not exist. Not even half of the functional proteins could survive without important function.
7. If two amino acids are located within the peptidyl transferase center, they will easily form a peptide bond. But as soon as you do that in the absence of the ribosome, the ends of the amino acids come together, forming a cyclic structure. Polymers cannot form. But if the ends are kept apart, by a theoretical primitive ribosome, a chain of peptide bonds could grow into a polymer.
8. An irreducibly complex system cannot be produced gradually by slight, successive modifications of a precursor system, since any precursor to an irreducibly complex system is by definition nonfunctional. Since natural selection requires a function to select, an irreducibly complex biological system, if there is such a thing, would have to arise as an integrated unit for natural selection to have anything to act on. It is almost universally conceded that such a sudden event would be irreconcilable with the gradualism Darwin envisioned.
9. This is creation by an intelligent designer, and this is the dictionary meaning of the word God.
The individual macromolecules are complex
But the complex interaction of biological macromolecules is only one aspect of the problem facing the origin of life. What compounds the enigma is that the individual macromolecular components are themselves complex, in the sense that their sequences - of ribonucleotides in the case of RNA, or amino acids for proteins - are very specific. The linear amino acid sequence of a protein is specific because it must (a) be able to fold into a discrete 3-dimensional structure, and (b) have the right amino acids in the right positions in the linear sequence so that, when folded, they are in exactly the right positions in relation to each other to form the active site(s) of the protein. (And similar considerations apply to RNAs.) Sequences which meet these criteria are exceedingly rare compared with the astronomical number of possible sequences of a suitable length. For example Douglas Axe has estimated that only 1 in about 10^74 possible sequences will have biological function (Axe). So it is totally unrealistic to think that such sequences could have arisen by chance. How much less a suite of mutually dependent macromolecules? If the components themselves were not so improbable then it might be realistic to think that a complex combination of components could arise by chance; but the extreme improbability of the individual components is such that they are very unlikely to arise individually, and hence there is no chance whatever of an interdependent system. 5
Where even just two macromolecules are required to perform a function, then it would be necessary for both components to arise together: Because natural selection does not have foresight: if one component arises alone it will not be retained for potential future usefulness (when the second component is available), but will almost certainly degrade by mutation. And, it should be noted, if the probability of getting one component is 1 in 10^74 then the probability of getting two together is 1 in 10^148 (not 1 in 2x10^74); and so on for multi-component systems. This is why the obligatory mutual dependence of many macromolecules in even basic biological systems completely defies any hope of an evolutionary origin. So, in summary, the crux of the problem is that even a basic biological replicating system requires (a) several macromolecules with complementary functions with (b) each having a highly improbable sequence. And this combination of complexities presents an insurmountable challenge to a naturalistic origin of life.
Proteins need to interact together. They need interface compatibility. So this adds to the unsurmountable problem ot making them by chance. Lets say our good old friend chance , after 10^78 trial and errors , got a functional protein made. That function has imho no value, if it cannot interact properly with other proteins. Then our same old friend made another 10^78 trial and errors attempts, and got the second protein made. How many attempts would be required to make them compatible to be able to interact in a functional way ? So lets suppose for a moment, that they got the right interface compatibility, and interact properly . So what ? This interaction alone has a long way to go, to get all 20 protein complexes needed to get DNA replication done, and making them interact all together in a functional way. Its the same as to make a piston by trial and error. At the end you have eventually a device that is piston like. If you do not know imho exactly the size the piston has to have to fit into the motor block, nothing done. Nothing will go. So FORSIGHT and INTELLIGENCE in order to PROJECT the whole device is ESSENTIAL.
The proteins in living cells are made of just certain kinds of amino acids, those that are “alpha” (short) and “left-handed.” Miller’s “primordial soup” contained many long (beta, gamma, delta) amino acids and equal numbers of both right-and left-handed forms. Problem: just one long or right-handed amino acid inserted into a chain of short, left-handed amino acids would prevent the coiling and folding necessary for proper protein function. What Miller actually produced was a seething brew of potent poisons that would absolutely destroy any hope for the chemical evolution of life. 1
Paper Reports that Amino Acids Used by Life Are Finely Tuned to Explore "Chemistry Space" 4
A recent paper in Nature's journal Scientific Reports, "Extraordinarily Adaptive Properties of the Genetically Encoded Amino Acids," 3 has found that the twenty amino acids used by life are finely tuned to explore "chemistry space" and allow for maximal chemical reactions. Considering that this is a technical paper, they give an uncommonly lucid and concise explanation of what they did:
We drew 108 random sets of 20 amino acids from our library of 1913 structures and compared their coverage of three chemical properties: size, charge, and hydrophobicity, to the standard amino acid alphabet. We measured how often the random sets demonstrated better coverage of chemistry space in one or more, two or more, or all three properties. In doing so, we found that better sets were extremely rare. In fact, when examining all three properties simultaneously, we detected only six sets with better coverage out of the 108 possibilities tested. That's quite striking: out of 100 million different sets of twenty amino acids that they measured, only six are better able to explore "chemistry space" than the twenty amino acids that life uses. That suggests that life's set of amino acids is finely tuned to one part in 16 million. Of course they only looked at three factors -- size, charge, and hydrophobicity. When we consider other properties of amino acids, perhaps our set will turn out to be the best:
While these three dimensions of property space are sufficient to demonstrate the adaptive advantage of the encoded amino acids, they are necessarily reductive and cannot capture all of the structural and energetic information contained in the 'better coverage' sets. They attribute this fine-tuning to natural selection, as their approach is to compare chance and selection as possible explanations of life's set of amino acids: This is consistent with the hypothesis that natural selection influenced the composition of the encoded amino acid alphabet, contributing one more clue to the much deeper and wider debate regarding the roles of chance versus predictability in the evolution of life.
But selection just means it is optimized and not random. They are only comparing two possible models -- selection and chance. They don't consider the fact that intelligent design is another cause that's capable of optimizing features. The question is: Which cause -- natural selection or intelligent design -- optimized this trait?
To do so, you'd have to consider the complexity required to incorporate a new amino acid into life's genetic code. That in turn would require lots of steps: a new codon to encode that amino acid, and new enzymes and RNAs to help process that amino acid during translation. In other words, incorporating a new amino acid into life's genetic code is a multimutation feature.
The biochemical language of the genetic code uses short strings of three nucleotides (called codons) to symbolize commands -- including start commands, stop commands, and codons that signify each of the 20 amino acids used in life. After the information in DNA is transcribed into mRNA, a series of codons in the mRNA molecule instructs the ribosome which amino acids are to be strung in which order to build a protein. Translation works by using another type of RNA molecule called transfer RNA (tRNA). During translation, tRNA molecules ferry needed amino acids to the ribosome so the protein chain can be assembled.
Each tRNA molecule is linked to a single amino acid on one end, and at the other end exposes three nucleotides (called an anti-codon). At the ribosome, small free-floating pieces of tRNA bind to the mRNA. When the anti-codon on a tRNA molecule binds to matching codons on the mRNA molecule at the ribosome, the amino acids are broken off the tRNA and linked up to build a protein.
For the genetic code to be translated properly, each tRNA molecule must be attached to the proper amino acid that corresponds to its anticodon as specified by the genetic code. If this critical step does not occur, then the language of the genetic code breaks down, and there is no way to convert the information in DNA into properly ordered proteins. So how do tRNA molecules become attached to the right amino acid?
Cells use special proteins called aminoacyl tRNA synthetase (aaRS) enzymes to attach tRNA molecules to the "proper" amino acid under thelanguage of the genetic code. Most cells use 20 different aaRS enzymes, one for each amino acid used in life. These aaRS enzymes are key to ensuring that the genetic code is correctly interpreted in the cell.
Yet these aaRS enzymes themselves are encoded by the genes in the DNA. This forms the essence of a "chicken-egg problem": aaRS enzymes themselves are necessary to perform the very task that constructs them.
How could such an integrated, language-based system arise in a step-by-step fashion? If any component is missing, the genetic information cannot be converted into proteins, and the message is lost. The RNA world is unsatisfactory because it provides no explanation for how the key step of the genetic code -- linking amino acids to the correct tRNA -- could have arisen.
Few of the many possible polypeptide chains will be useful to Cells
Paper Reports that Amino Acids Used by Life Are Finely Tuned to Explore "Chemistry Space" 4
A recent paper in Nature's journal Scientific Reports, "Extraordinarily Adaptive Properties of the Genetically Encoded Amino Acids," 3 has found that the twenty amino acids used by life are finely tuned to explore "chemistry space" and allow for maximal chemical reactions. Considering that this is a technical paper, they give an uncommonly lucid and concise explanation of what they did:
We drew 108 random sets of 20 amino acids from our library of 1913 structures and compared their coverage of three chemical properties: size, charge, and hydrophobicity, to the standard amino acid alphabet. We measured how often the random sets demonstrated better coverage of chemistry space in one or more, two or more, or all three properties. In doing so, we found that better sets were extremely rare. In fact, when examining all three properties simultaneously, we detected only six sets with better coverage out of the 108 possibilities tested. That's quite striking: out of 100 million different sets of twenty amino acids that they measured, only six are better able to explore "chemistry space" than the twenty amino acids that life uses. That suggests that life's set of amino acids is finely tuned to one part in 16 million. Of course they only looked at three factors -- size, charge, and hydrophobicity. When we consider other properties of amino acids, perhaps our set will turn out to be the best:
While these three dimensions of property space are sufficient to demonstrate the adaptive advantage of the encoded amino acids, they are necessarily reductive and cannot capture all of the structural and energetic information contained in the 'better coverage' sets. They attribute this fine-tuning to natural selection, as their approach is to compare chance and selection as possible explanations of life's set of amino acids: This is consistent with the hypothesis that natural selection influenced the composition of the encoded amino acid alphabet, contributing one more clue to the much deeper and wider debate regarding the roles of chance versus predictability in the evolution of life.
But selection just means it is optimized and not random. They are only comparing two possible models -- selection and chance. They don't consider the fact that intelligent design is another cause that's capable of optimizing features. The question is: Which cause -- natural selection or intelligent design -- optimized this trait?
To do so, you'd have to consider the complexity required to incorporate a new amino acid into life's genetic code. That in turn would require lots of steps: a new codon to encode that amino acid, and new enzymes and RNAs to help process that amino acid during translation. In other words, incorporating a new amino acid into life's genetic code is a multimutation feature.
The biochemical language of the genetic code uses short strings of three nucleotides (called codons) to symbolize commands -- including start commands, stop commands, and codons that signify each of the 20 amino acids used in life. After the information in DNA is transcribed into mRNA, a series of codons in the mRNA molecule instructs the ribosome which amino acids are to be strung in which order to build a protein. Translation works by using another type of RNA molecule called transfer RNA (tRNA). During translation, tRNA molecules ferry needed amino acids to the ribosome so the protein chain can be assembled.
Each tRNA molecule is linked to a single amino acid on one end, and at the other end exposes three nucleotides (called an anti-codon). At the ribosome, small free-floating pieces of tRNA bind to the mRNA. When the anti-codon on a tRNA molecule binds to matching codons on the mRNA molecule at the ribosome, the amino acids are broken off the tRNA and linked up to build a protein.
For the genetic code to be translated properly, each tRNA molecule must be attached to the proper amino acid that corresponds to its anticodon as specified by the genetic code. If this critical step does not occur, then the language of the genetic code breaks down, and there is no way to convert the information in DNA into properly ordered proteins. So how do tRNA molecules become attached to the right amino acid?
Cells use special proteins called aminoacyl tRNA synthetase (aaRS) enzymes to attach tRNA molecules to the "proper" amino acid under thelanguage of the genetic code. Most cells use 20 different aaRS enzymes, one for each amino acid used in life. These aaRS enzymes are key to ensuring that the genetic code is correctly interpreted in the cell.
Yet these aaRS enzymes themselves are encoded by the genes in the DNA. This forms the essence of a "chicken-egg problem": aaRS enzymes themselves are necessary to perform the very task that constructs them.
How could such an integrated, language-based system arise in a step-by-step fashion? If any component is missing, the genetic information cannot be converted into proteins, and the message is lost. The RNA world is unsatisfactory because it provides no explanation for how the key step of the genetic code -- linking amino acids to the correct tRNA -- could have arisen.
Few of the many possible polypeptide chains will be useful to Cells
Paul Davies puts it more graphically: ‘Making a protein simply by injecting energy is rather like exploding a stick of dynamite under a pile of bricks and expecting it to form a house. You may liberate enough energy to raise the bricks, but without coupling the energy to the bricks in a controlled and ordered way, there is little hope of producing anything other than a chaotic mess.’ It is one thing to produce bricks; it is an entirely different thing to organize the building of a house or factory. If you had to, you could build a house using stones that you found lying around, in all the shapes and sizes in which they came due to natural causes. However, the organization of the building requires something that is not contained in the stones. It requires the intelligence of the architect and the skill of the builder. It is the same with the building blocks of life. Blind chance just will not do the job of putting them together in a specific way. Organic chemist and molecular biologist A.G. Cairns-Smith puts it this way: ‘Blind chance… is very limited… he can produce exceedingly easily the equivalent of letters and small words, but he becomes very quickly incompetent as the amount of organization increases. Very soon indeed long waiting periods and massive material resources become irrelevant.’
Bruce Alberts writes in Molecular biology of the cell :
Since each of the 20 amino acids is chemically distinct and each can, in principle, occur at any position in a protein chain, there are 20 x 20 x 20 x 20 = 160,000 different possible polypeptide chains four amino acids long, or 20n different possible polypeptide chains n amino acids long. For a typical protein length of about 300 amino acids, a cell could theoretically make more than 10^390 different pollpeptide chains. This is such an enormous number that to produce just one molecule of each kind would require many more atoms than exist in the universe. Only a very small fraction of this vast set of conceivable polypeptide chains would adopt a single, stable three-dimensional conformation-by some estimates, less than one in a billion. And yet the vast majority of proteins present in cells adopt unique and stable conformations. How is this possible?
The complexity of living organisms is staggering, and it is quite sobering to note that we currently lack even the tiniest hint of what the function might be for more than 10,000 of the proteins that have thus far been identified in the human genome. There are certainly enormous challenges ahead for the next generation of cell biologists, with no shortage of fascinating mysteries to solve.
Now comes Alberts striking explanation of how the right sequence arised :
The answer Iies in natural selection. A protein with an unpredictably variable structure and biochemical activity is unlikely to help the survival of a cell that contains it. Such
proteins would therefore have been eliminated by natural selection through the enormously long trial-and-error process that underlies biological evolution. Because evolution has selected for protein function in living organisms, the amino acid sequence of most present-day proteins is such that a single conformation is extremely stable. In addition, this conformation has its chemical properties finely tuned to enable the protein to perform a particular catalltic or structural function in the cell. Proteins are so precisely built that the change of even a few atoms in one amino acid can sometimes disrupt the structure of the whole molecule so severelv that all function is lost.
Proteins are not rigid lumps of material. They often have precisely engineered moving parts whose mechanical actions are coupled to chemical events. It is this coupling of chemistry and movement that gives proteins the extraordinary capabilities that underlie the dynamic processes in living cells
Now think for a moment . It seems that natural selection is the key answer to any phenomena in biology, where there is no scientific evidence to make an empirical claim. Much has been written about the fact that natural selection cannot produce coded information. Alberts's short explanation is a prima facie example of how mainstream scientists make without hesitation " just so " claims without being able to provide a shred of evidence, just in order to maintain a paradigm on which the scientific establishment relies, where evolution is THE answer to almost every biochemical phenomena. The fact is that precision, coded information, stability, interdependence and irreducible complexity, etc. are products of intelligent minds. The author seems also to forget that natural selection cannot occur before the first living cell replicates. Several hundred proteins had to be already in place and fully operating in order to make even the simplest life possible
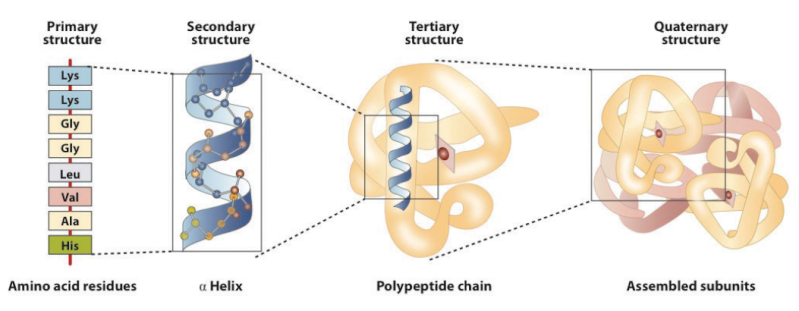
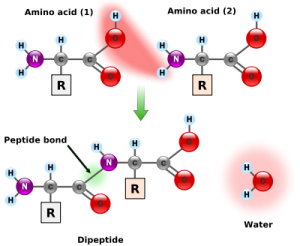
Amino acids link together when the amino group of one amino acid bonds to the carboxyl group of another. Notice that water is a by-product of the reaction (called a condensation reaction).
Stephen Meyer writes in Signature of the cell:
According to neo-Darwinian theory, new genetic information arises first as random mutations occur in the DNA of existing organisms. When mutations arise that confer a survival advantage on the organisms that possess them, the resulting genetic changes are passed on by natural selection to the next generation. As these changes accumulate, the features of a population begin to change over time. Nevertheless, natural selection can "select" only what random mutations first produce. And for the evolutionary process to produce new forms of life, random mutations must first have produced new genetic information for building novel proteins. That, for the
mathematicians, physicists, and engineers at Wistar, was the problem. Why?
The skeptics at Wistar argued that it is extremely difficult to assemble a new gene or protein by chance because of the sheer number of possible base or amino-acid sequences. For every combination of amino acids that produces a functional protein there exists a vast number of other possible combinations that do not. And as the length of the required protein grows, the number of possible amino-acid sequence combinations of that length grows exponentially, so that the odds of finding a functional sequence—that is, a working protein—diminish precipitously.
To see this, consider the following. Whereas there are four ways to combine the letters A and B to make a two-letter combination (AB, BA, AA, and BB), there are eight ways to make three-letter combinations (AAA, AAB, ABB, ABA, BAA, BBA, BAB, BBB), and sixteen ways to make four-letter combinations, and so on. The number of combinations grows geometrically, 22, 23, 24, and so on. And this growth becomes more pronounced when the set of letters is larger. For protein chains, there are 202, or 400, ways to make a two-amino-acid combination, since each position could be any one of 20 different alphabetic characters. Similarly, there are 203, or 8,000, ways to make a three-amino-acid sequence, and 204, or 160,000, ways to make a sequence four amino acids long, and so on. As the number of possible combinations rises, the odds of finding a correct sequence diminishes correspondingly. But most functional proteins are made of hundreds of amino acids. Therefore, even a relatively short protein of, say, 150 amino acids represents one sequence among an astronomically large number of other possible sequence combinations (approximately 10^195).
Consider the way this combinatorial problem might play itself out in the case of proteins in a hypothetical prebiotic soup. To construct even one short protein molecule of 150 amino acids by chance within the prebiotic soup there are several combinatorial problems—probabilistic hurdles—to overcome. First, all amino acids must form a chemical bond known as a peptide bond when joining with other amino acids in the protein chain
Consider the way this combinatorial problem might play itself out in the case of proteins in a hypothetical prebiotic soup. To construct even one short protein molecule of 150 amino acids by chance within the prebiotic soup there are several combinatorial problems—probabilistic hurdles—to overcome. First, all amino acids must form a chemical bond known as a peptide bond when joining with other amino acids in the protein chain (see Fig. 9.1). If the amino acids do not link up with one another via a peptide bond, the resulting molecule will not fold into a protein. In nature many other types of chemical bonds are possible between amino acids. In fact, when amino-acid mixtures are allowed to react in a test tube, they form peptide and nonpeptide bonds with roughly equal probability. Thus, with each amino-acid addition, the probability of it forming a peptide bond is roughly 1/2. Once four amino acids have become linked, the likelyhood that they are joined exclusively by peptide bonds is roughly 1/2 × 1/2 × 1/2 ×
1/2 = 1/16, or (1/2)4. The probability of building a chain of 150 amino acids in which all linkages are peptide linkages is (1/2)149, or roughly 1 chance in 10^45.
Second, in nature every amino acid found in proteins (with one exception) has a distinct mirror image of itself; there is one left-handed version, or L-form, and one right-handed version, or D-form. These mirror-image forms are called optical isomers (see Fig. 9.2). Functioning proteins tolerate only left-handed amino acids, yet in abiotic amino-acid production the right-handed and left-handed isomers are produced with roughly equal frequency. Taking this into consideration further compounds the improbability of attaining a biologically functioning protein. The probability of attaining, at random, only L-amino acids in a hypothetical peptide chain 150 amino acids long is (1/2)150, or again roughly 1 chance in 1045. Starting from mixtures of D-forms and L-forms, the probability of building a 150-amino-acid chain at random in which all bonds are peptide bonds and all amino acids are L-form is, therefore, roughly 1 chance in 1090.
Second, in nature every amino acid found in proteins (with one exception) has a distinct mirror image of itself; there is one left-handed version, or L-form, and one right-handed version, or D-form. These mirror-image forms are called optical isomers . Functioning proteins tolerate only left-handed amino acids, yet in abiotic amino-acid production the right-handed and left-handed isomers are produced with roughly equal frequency. Taking this into consideration further compounds the improbability of attaining a biologically functioning protein. The probability of attaining, at random, only L-amino acids in a hypothetical peptide chain 150 amino acids long is (1/2)150, or again roughly 1 chance in 10^45. Starting from mixtures of D-forms and L-forms, the probability of building a 150-amino-acid chain at random in which all bonds are peptide bonds and all amino acids are L-form is, therefore, roughly 1 chance in 10^90.
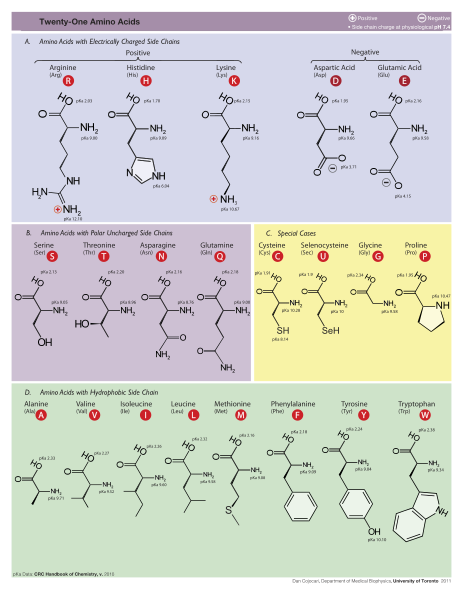
Functioning proteins have a third independent requirement, the most important of all: their amino acids, like letters in a meaningful sentence, must link up in functionally specified sequential arrangements. In some cases, changing even one amino acid at a given site results in the loss of protein function. Moreover, because there are 20 biologically occurring amino acids, the probability of getting a specific amino acid at a given site is small—1/20. (Actually the probability is even lower because, in nature, there are also many nonprotein-forming amino acids.) On the assumption that each site in a protein chain requires a particular amino acid, the probability of attaining a particular protein 150 amino acids long would be (1/20)150, or roughly 1 chance in 10^195.
How rare, or common, are the functional sequences of amino acids among all the possible sequences of amino acids in a chain of any given length?
Douglas Axe answered this question in 2004 3 , and Axe was able to make a careful estimate of the ratio of (a) the number of 150-amino-acid sequences that can perform that particular function to (b) the whole set of possible amino-acid sequences of this length. Axe estimated this ratio to be 1 to 10^77.
This was a staggering number, and it suggested that a random process would have great difficulty generating a protein with that particular function by chance. But I didn't want to know just the likelihood of finding a protein with a particular function within a space of combinatorial possibilities. I wanted to know the odds of finding any functional protein whatsoever within such a space. That number would make it possible to evaluate chance-based origin-of-life scenarios, to assess the probability that a single protein—any working protein—would have arisen by chance on the early earth.
Fortunately, Axe's work provided this number as well.17 Axe knew that in nature proteins perform many specific functions. He also knew that in order to perform these functions their amino-acid chains must first fold into stable three-dimensional structures. Thus, before he estimated the frequency of sequences performing a specific (beta-lactamase) function, he first performed experiments that enabled him to estimate the frequency of sequences that will produce stable folds. On the basis of his experimental results, he calculated the ratio of (a) the number of 150-amino-acid sequences capable of folding into stable "function-ready" structures to (b) the whole set of possible amino-acid sequences of that length. He determined that ratio to be 1 to 10^74.
In other words, a random process producing amino-acid chains of this length would stumble onto a functional protein only about once in every 10^74 attempts.
When one considers that Robert Sauer was working on a shorter protein of 100 amino acids, Axe's number might seem a bit less prohibitively improbable. Nevertheless, it still represents a startlingly small probability. In conversations with me, Axe has compared the odds of producing a functional protein sequence of modest (150-amino-acid) length at random to the odds of finding a single marked atom out of all the atoms in our galaxy via a blind and undirected search. Believe it or not, the odds of finding the marked atom in our galaxy are markedly better (about a billion times better) than those of finding a functional protein among all the sequences of corresponding length.
Problems with Making Mutation the Basis for MacroevolutionBruce Alberts writes in Molecular biology of the cell :
Since each of the 20 amino acids is chemically distinct and each can, in principle, occur at any position in a protein chain, there are 20 x 20 x 20 x 20 = 160,000 different possible polypeptide chains four amino acids long, or 20n different possible polypeptide chains n amino acids long. For a typical protein length of about 300 amino acids, a cell could theoretically make more than 10^390 different pollpeptide chains. This is such an enormous number that to produce just one molecule of each kind would require many more atoms than exist in the universe. Only a very small fraction of this vast set of conceivable polypeptide chains would adopt a single, stable three-dimensional conformation-by some estimates, less than one in a billion. And yet the vast majority of proteins present in cells adopt unique and stable conformations. How is this possible?
The complexity of living organisms is staggering, and it is quite sobering to note that we currently lack even the tiniest hint of what the function might be for more than 10,000 of the proteins that have thus far been identified in the human genome. There are certainly enormous challenges ahead for the next generation of cell biologists, with no shortage of fascinating mysteries to solve.
Now comes Alberts striking explanation of how the right sequence arised :
The answer Iies in natural selection. A protein with an unpredictably variable structure and biochemical activity is unlikely to help the survival of a cell that contains it. Such
proteins would therefore have been eliminated by natural selection through the enormously long trial-and-error process that underlies biological evolution. Because evolution has selected for protein function in living organisms, the amino acid sequence of most present-day proteins is such that a single conformation is extremely stable. In addition, this conformation has its chemical properties finely tuned to enable the protein to perform a particular catalltic or structural function in the cell. Proteins are so precisely built that the change of even a few atoms in one amino acid can sometimes disrupt the structure of the whole molecule so severelv that all function is lost.
Proteins are not rigid lumps of material. They often have precisely engineered moving parts whose mechanical actions are coupled to chemical events. It is this coupling of chemistry and movement that gives proteins the extraordinary capabilities that underlie the dynamic processes in living cells
Now think for a moment . It seems that natural selection is the key answer to any phenomena in biology, where there is no scientific evidence to make an empirical claim. Much has been written about the fact that natural selection cannot produce coded information. Alberts's short explanation is a prima facie example of how mainstream scientists make without hesitation " just so " claims without being able to provide a shred of evidence, just in order to maintain a paradigm on which the scientific establishment relies, where evolution is THE answer to almost every biochemical phenomena. The fact is that precision, coded information, stability, interdependence and irreducible complexity, etc. are products of intelligent minds. The author seems also to forget that natural selection cannot occur before the first living cell replicates. Several hundred proteins had to be already in place and fully operating in order to make even the simplest life possible

Amino acids link together when the amino group of one amino acid bonds to the carboxyl group of another. Notice that water is a by-product of the reaction (called a condensation reaction).
Stephen Meyer writes in Signature of the cell:
According to neo-Darwinian theory, new genetic information arises first as random mutations occur in the DNA of existing organisms. When mutations arise that confer a survival advantage on the organisms that possess them, the resulting genetic changes are passed on by natural selection to the next generation. As these changes accumulate, the features of a population begin to change over time. Nevertheless, natural selection can "select" only what random mutations first produce. And for the evolutionary process to produce new forms of life, random mutations must first have produced new genetic information for building novel proteins. That, for the
mathematicians, physicists, and engineers at Wistar, was the problem. Why?
The skeptics at Wistar argued that it is extremely difficult to assemble a new gene or protein by chance because of the sheer number of possible base or amino-acid sequences. For every combination of amino acids that produces a functional protein there exists a vast number of other possible combinations that do not. And as the length of the required protein grows, the number of possible amino-acid sequence combinations of that length grows exponentially, so that the odds of finding a functional sequence—that is, a working protein—diminish precipitously.
To see this, consider the following. Whereas there are four ways to combine the letters A and B to make a two-letter combination (AB, BA, AA, and BB), there are eight ways to make three-letter combinations (AAA, AAB, ABB, ABA, BAA, BBA, BAB, BBB), and sixteen ways to make four-letter combinations, and so on. The number of combinations grows geometrically, 22, 23, 24, and so on. And this growth becomes more pronounced when the set of letters is larger. For protein chains, there are 202, or 400, ways to make a two-amino-acid combination, since each position could be any one of 20 different alphabetic characters. Similarly, there are 203, or 8,000, ways to make a three-amino-acid sequence, and 204, or 160,000, ways to make a sequence four amino acids long, and so on. As the number of possible combinations rises, the odds of finding a correct sequence diminishes correspondingly. But most functional proteins are made of hundreds of amino acids. Therefore, even a relatively short protein of, say, 150 amino acids represents one sequence among an astronomically large number of other possible sequence combinations (approximately 10^195).
Consider the way this combinatorial problem might play itself out in the case of proteins in a hypothetical prebiotic soup. To construct even one short protein molecule of 150 amino acids by chance within the prebiotic soup there are several combinatorial problems—probabilistic hurdles—to overcome. First, all amino acids must form a chemical bond known as a peptide bond when joining with other amino acids in the protein chain
Consider the way this combinatorial problem might play itself out in the case of proteins in a hypothetical prebiotic soup. To construct even one short protein molecule of 150 amino acids by chance within the prebiotic soup there are several combinatorial problems—probabilistic hurdles—to overcome. First, all amino acids must form a chemical bond known as a peptide bond when joining with other amino acids in the protein chain (see Fig. 9.1). If the amino acids do not link up with one another via a peptide bond, the resulting molecule will not fold into a protein. In nature many other types of chemical bonds are possible between amino acids. In fact, when amino-acid mixtures are allowed to react in a test tube, they form peptide and nonpeptide bonds with roughly equal probability. Thus, with each amino-acid addition, the probability of it forming a peptide bond is roughly 1/2. Once four amino acids have become linked, the likelyhood that they are joined exclusively by peptide bonds is roughly 1/2 × 1/2 × 1/2 ×
1/2 = 1/16, or (1/2)4. The probability of building a chain of 150 amino acids in which all linkages are peptide linkages is (1/2)149, or roughly 1 chance in 10^45.
Second, in nature every amino acid found in proteins (with one exception) has a distinct mirror image of itself; there is one left-handed version, or L-form, and one right-handed version, or D-form. These mirror-image forms are called optical isomers (see Fig. 9.2). Functioning proteins tolerate only left-handed amino acids, yet in abiotic amino-acid production the right-handed and left-handed isomers are produced with roughly equal frequency. Taking this into consideration further compounds the improbability of attaining a biologically functioning protein. The probability of attaining, at random, only L-amino acids in a hypothetical peptide chain 150 amino acids long is (1/2)150, or again roughly 1 chance in 1045. Starting from mixtures of D-forms and L-forms, the probability of building a 150-amino-acid chain at random in which all bonds are peptide bonds and all amino acids are L-form is, therefore, roughly 1 chance in 1090.
Second, in nature every amino acid found in proteins (with one exception) has a distinct mirror image of itself; there is one left-handed version, or L-form, and one right-handed version, or D-form. These mirror-image forms are called optical isomers . Functioning proteins tolerate only left-handed amino acids, yet in abiotic amino-acid production the right-handed and left-handed isomers are produced with roughly equal frequency. Taking this into consideration further compounds the improbability of attaining a biologically functioning protein. The probability of attaining, at random, only L-amino acids in a hypothetical peptide chain 150 amino acids long is (1/2)150, or again roughly 1 chance in 10^45. Starting from mixtures of D-forms and L-forms, the probability of building a 150-amino-acid chain at random in which all bonds are peptide bonds and all amino acids are L-form is, therefore, roughly 1 chance in 10^90.
Functioning proteins have a third independent requirement, the most important of all: their amino acids, like letters in a meaningful sentence, must link up in functionally specified sequential arrangements. In some cases, changing even one amino acid at a given site results in the loss of protein function. Moreover, because there are 20 biologically occurring amino acids, the probability of getting a specific amino acid at a given site is small—1/20. (Actually the probability is even lower because, in nature, there are also many nonprotein-forming amino acids.) On the assumption that each site in a protein chain requires a particular amino acid, the probability of attaining a particular protein 150 amino acids long would be (1/20)150, or roughly 1 chance in 10^195.
How rare, or common, are the functional sequences of amino acids among all the possible sequences of amino acids in a chain of any given length?
Douglas Axe answered this question in 2004 3 , and Axe was able to make a careful estimate of the ratio of (a) the number of 150-amino-acid sequences that can perform that particular function to (b) the whole set of possible amino-acid sequences of this length. Axe estimated this ratio to be 1 to 10^77.
This was a staggering number, and it suggested that a random process would have great difficulty generating a protein with that particular function by chance. But I didn't want to know just the likelihood of finding a protein with a particular function within a space of combinatorial possibilities. I wanted to know the odds of finding any functional protein whatsoever within such a space. That number would make it possible to evaluate chance-based origin-of-life scenarios, to assess the probability that a single protein—any working protein—would have arisen by chance on the early earth.
Fortunately, Axe's work provided this number as well.17 Axe knew that in nature proteins perform many specific functions. He also knew that in order to perform these functions their amino-acid chains must first fold into stable three-dimensional structures. Thus, before he estimated the frequency of sequences performing a specific (beta-lactamase) function, he first performed experiments that enabled him to estimate the frequency of sequences that will produce stable folds. On the basis of his experimental results, he calculated the ratio of (a) the number of 150-amino-acid sequences capable of folding into stable "function-ready" structures to (b) the whole set of possible amino-acid sequences of that length. He determined that ratio to be 1 to 10^74.
In other words, a random process producing amino-acid chains of this length would stumble onto a functional protein only about once in every 10^74 attempts.
When one considers that Robert Sauer was working on a shorter protein of 100 amino acids, Axe's number might seem a bit less prohibitively improbable. Nevertheless, it still represents a startlingly small probability. In conversations with me, Axe has compared the odds of producing a functional protein sequence of modest (150-amino-acid) length at random to the odds of finding a single marked atom out of all the atoms in our galaxy via a blind and undirected search. Believe it or not, the odds of finding the marked atom in our galaxy are markedly better (about a billion times better) than those of finding a functional protein among all the sequences of corresponding length.
Dembsi, the design of life, general notes, page 11:
If the proportion of gene sequences that are biologically useful were large, there might be reason to think that point or chromosome mutations could be helpful in achieving the novel biological structures required by macroevolution. But all the evidence points to biologically useful gene sequences being exceedingly rare. It’s therefore highly unlikely that point and chromosome mutations can transform a duplicated gene into a novel functional gene. Genetic sequence space (i.e., the set of all possible genetic sequences) is functionally sparse (i.e., the overwhelming majority of genetic sequences don’t, and indeed can’t, do anything biologically useful or significant). As a consequence, navigating genetic sequence space by undirected means is no help getting from one island of functionality (i.e., one region of biologically useful or significant genetic sequences) to the next.
For instance, there is no evidence that conventional evolutionary mechanisms, such as natural selection, can evolve a gene in one region of genetic sequence space with one set of functions into a gene in a far distant region of genetic sequence space with another set of functions (distance here being measured in terms of sequence similarity). In the language of mathematical biology, genetic sequence space gives no indication of being highly interconnected by functional pathways that continuously connect genes with one function to genes with another (which would be required if natural selection, say, were to assist a duplicated gene in transforming into a novel gene). But there are still more problems with trying to make mutation the basis for macroevolution. For point and chromosome mutations to account for macroevolutionary change, it is not enough for individual genes to be transformed into novel genes that exhibit novel functions. Rather it is required that a whole suite of novel genes be produced through the coordinated transformation of existing genes. This is because for new biological structures to evolve (as required by macroevolution), many genes will have to change.
But it has not been demonstrated that mutations can produce the highly coordinated protein parts required for many biological structures. These are the structures that macroevolution would need to produce. Till now, however, there is no evidence for the coordinated macromutations required for macroevolution. The closest evidence cited in
textbooks includes increased immunity to malaria associated with the mutation for sickle-cell anemia and the resistance to antibiotics by mutant strains of bacteria.
In no such case, however, do we see a coordinated set of mutations that lead to complex novel structures. Sickle-cell anemia, for instance, is induced by a single point mutation that leads to single change in an amino acid (a valine is substituted for a glutamic acid in a hemoglobin molecule). Point mutations like this might enable organisms to stabilize and maintain themselves in the face of severe environmental pressure. In most instances, however, novel traits induced by such mutations do not continue to benefit the organisms when the environmental pressure is removed. Apart from environmental pressure, such mutations can even be deleterious. For instance, when an individual is heterozygous for sickle-cell anemia, the mutation provides an advantage for surviving the threat of malaria. It does so, however, at the expense of inflicting on homozygous individuals an anemia that impairs the transport of oxygen to the body’s cells. Indeed, sickle-cell anemia is often lethal.
To observe the origin of new species, one would expect to find it most readily in bacteria. That’s because bacteria can be easily mutated with chemicals and radiation in the laboratory, they take up very little space, and they have very short generation times. Indeed, thousands of mutations, billions of organisms, and thousands of generations can be studied by a single scientist. Yet, bacteriologists have never witnessed the origin of a new species. (Some new plant species have been observed to originate through hybridization, but the combining of two species to make a third is the opposite of the Darwinian process of splitting one species into two.) For mutations to contribute to evolution, they must benefit the organism. If a mutation harms the organism, it will tend to be eliminated, rather than favored, by natural selection. The only beneficial mutations that have ever been observed, in bacteria or in any other kind of organism, have been biochemical. That is, they affect only single molecules (such as the target molecule for streptomycin). There are no known beneficial mutations affecting morphology, or shape. All known morphological mutations are either neutral (i.e., they don’t have any noticeable effect on the organism’s fitness), or they are harmful—and the bigger their effect the more harmful they are. Yet, Darwinian evolution (i.e., the origin of new species, new organs, and new body plans) clearly requires changes in shape. So, there is no evidence for (and indeed a lot of evidence against) a role for mutations as providing raw materials for Darwinian evolution.
The specific genetic changes that give rise to the evolutionary origins of novel protein-protein interactions have rarely been documented in detail 6
Although numerous investigators assume that the global features of genetic networks are moulded by natural selection, there has been no formal demonstration of the adaptive origin of any genetic network 7 The mechanisms by which genetic networks become established evolutionarily are far from clear.
Many physicists, engineers and computer scientists, and some cell and developmental biologists, are convinced that biological networks exhibit properties that could only be products of natural selection; however, the matter has rarely been examined in the context of well-established evolutionary principles.
Alon states that it is “…wondrous that the solutions found by evolution have much in common with good engineering,”
Biological designs require a great deal more than the specification of a single DNA residue. Proteins, for instance, require a great many DNA residues to be specified. Even a single, typical protein requires more than 10^100 (a one followed by one hundred zeros) evolutionary experiments to find. This is an astronomical problem that is well beyond evolution’s capabilities. See here, here, here and here for more details. [url=10. http://darwins-god.blogspot.com.br/2011/06/vision-cascade-is-initiated-not-by.html]8[/url]
1) https://answersingenesis.org/origin-of-life/the-origin-of-life-dna-and-protein/
2) B.Alberts Molecular biology of the cell.
3) http://www.ncbi.nlm.nih.gov/pubmed/15321723
4) http://www.evolutionnews.org/2015/06/paper_reports_t096581.html
5) https://www.c4id.org.uk/index.php?option=com_content&view=article&id=211:the-problem-of-the-origin-of-life&catid=50:genetics&Itemid=43
6. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0001054
7. https://sci-hub.bz/http://www.nature.com/nrg/journal/v8/n10/abs/nrg2192.html
8. http://darwins-god.blogspot.com.br/2011/06/vision-cascade-is-initiated-not-by.html
9. Molecular Mechanisms of Autonomy in Biological Systems Relativity of Code, Energy and Mass, page 34
10. https://www.nasa.gov/sites/default/files/atoms/files/pohorille_wilson_shannon_2017_1.pdf
More readings:
Comprehensive experimental fitness landscape and evolutionary network for small RNA
Our study suggests that replaying the “tape of life” at the very origin of life might lead to quite different results.
Last edited by Otangelo on Wed Nov 09, 2022 6:28 am; edited 42 times in total