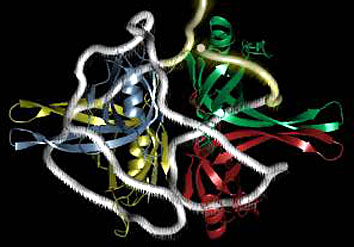
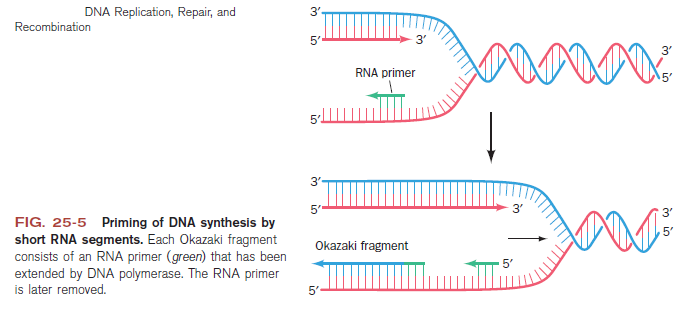
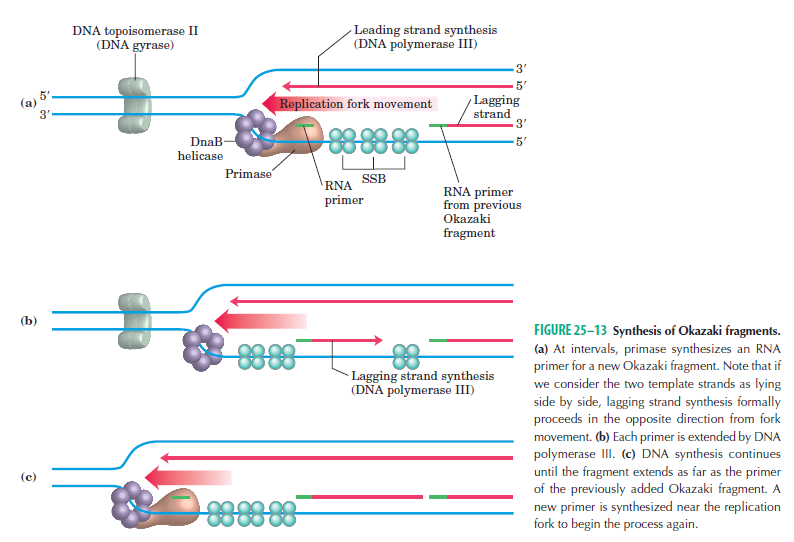
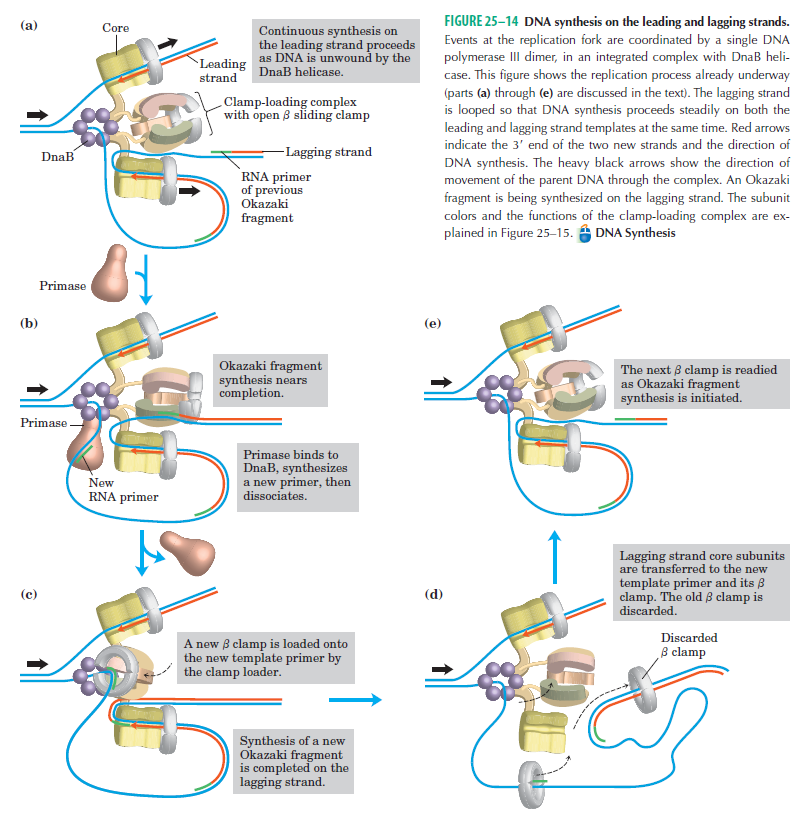
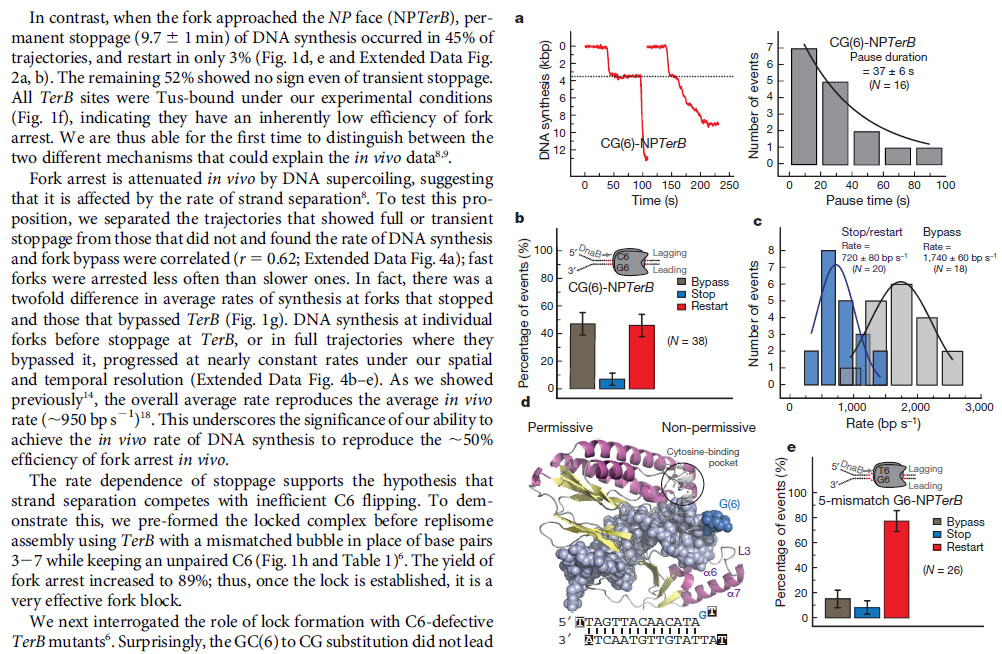
The Hexameric DnaB Helicase
DNA HELICASES: UNWINDING OF THE DOUBLE HELIX
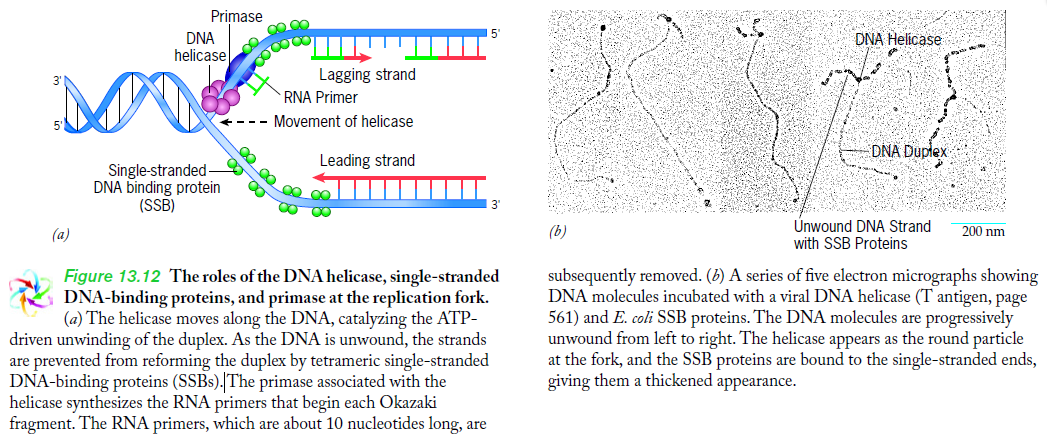
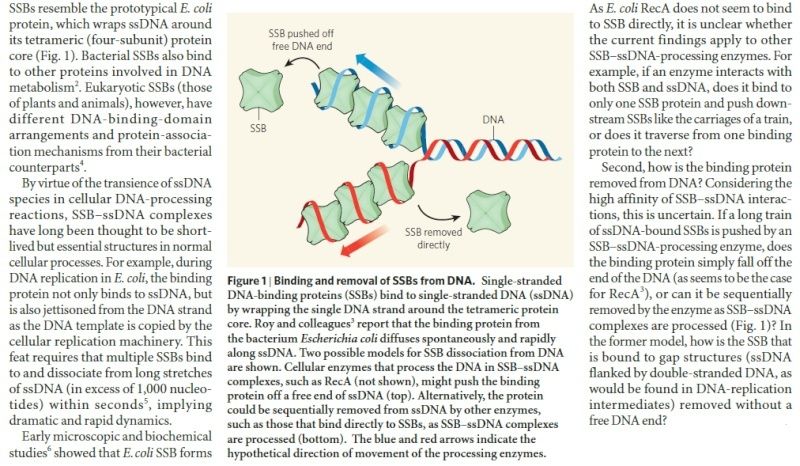
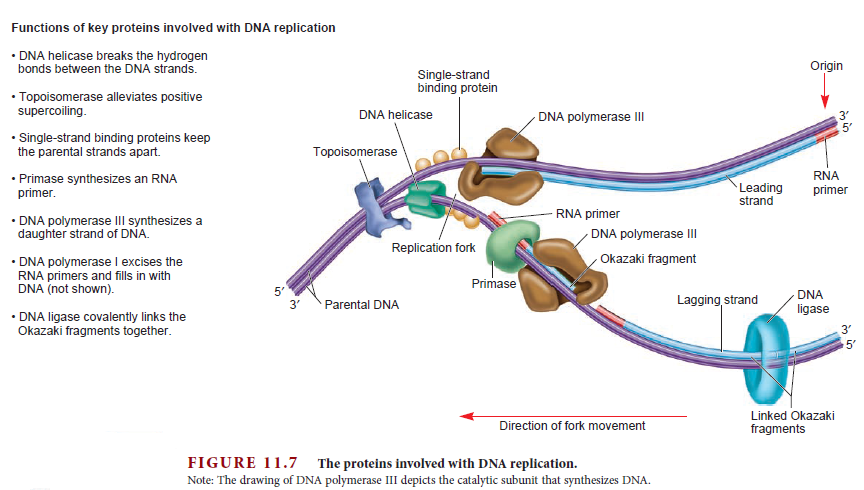
DNA helicase unwinds double-stranded DNA for copying
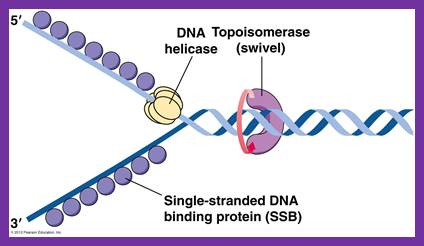
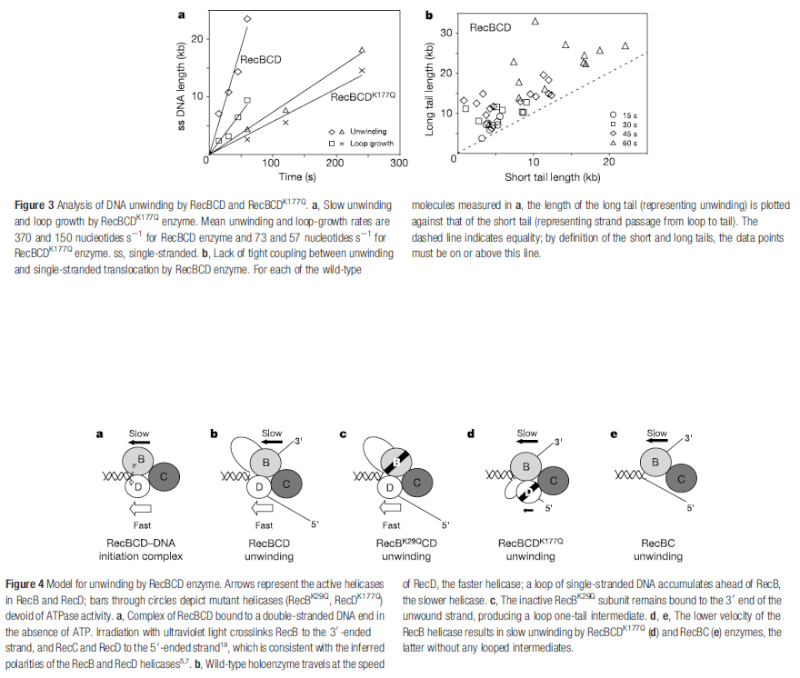
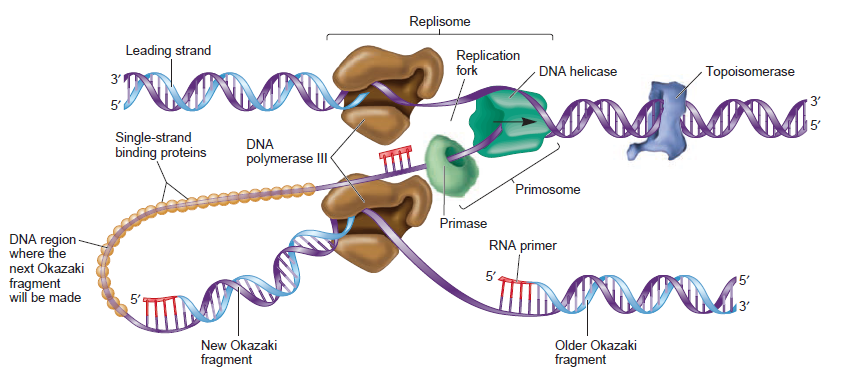
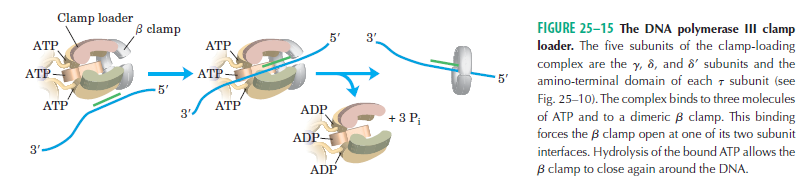
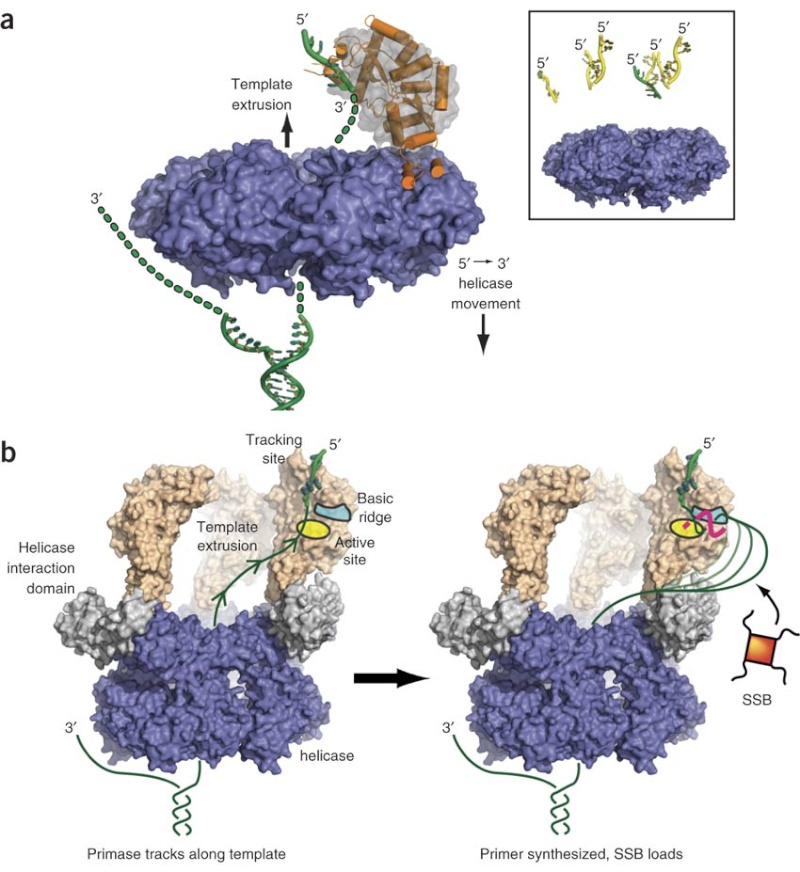
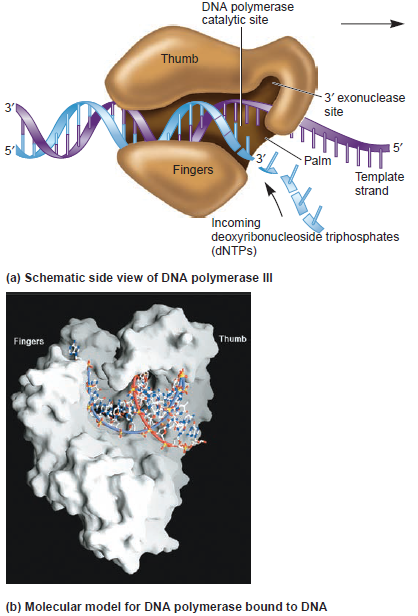
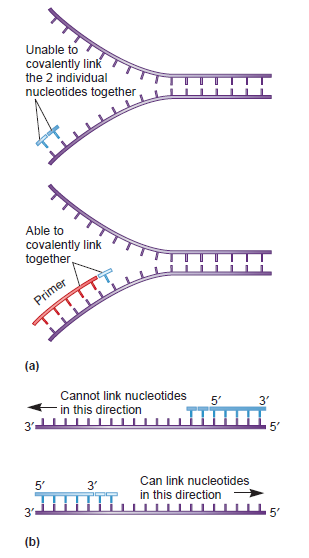
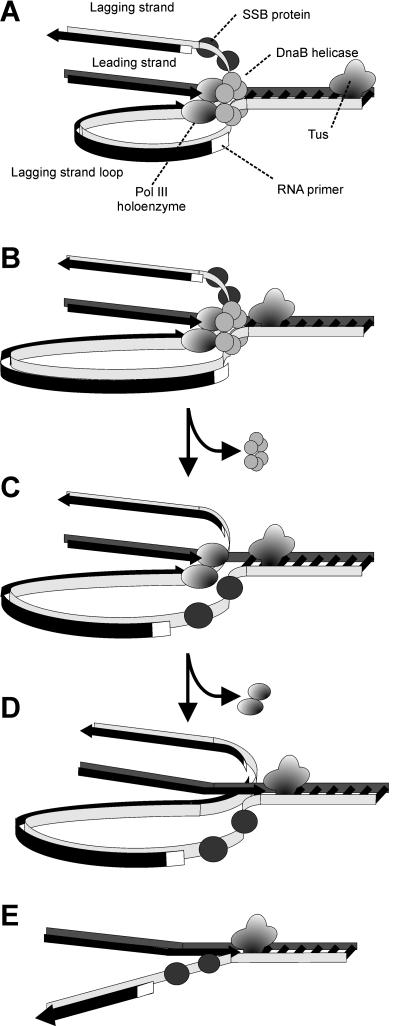
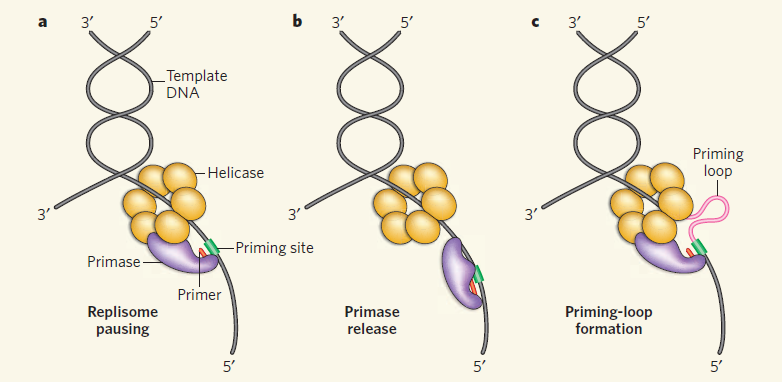
Having now considered the key enzymes involved in the actual synthesis of DNA during replication, let us now turn to an enzyme that has an important function before synthesis can actually occur – the unwinding of the double-stranded helix of the parent DNA. When base-paired in the double-stranded helical DNA molecule, nucleotides are inaccessible to polymerases, making copying impossible. Both primase and polymerases need access to single-stranded DNA. To achieve this, the internal base-pairing must be broken and the helix unwound. The first step in this unwinding process is the initial opening of the helix, a step performed by the initiator protein at the origin of replication. Once opened, the unwinding of the double helix to expose single-stranded DNA for copying can begin. This unwinding process is catalyzed by an enzyme called DNA helicase. The cell also needs to open the helix for DNA repair and recombination, and there are a unique set of DNA helicases for this particular case of DNA unwinding. Helicases open the double-stranded DNA and then travel with the replication fork, continuously unwinding the DNA to provide a template for the polymerase to copy. The helicases involved in replication in both bacteria and eukaryotes are composed of six subunits that form a ring structure that surrounds one strand of the DNA. The structure of the replicative DNA helicase from papillomavirusprovides insights into the mechanism of helicase action. This viral helicase is a hexamer of one protein, E1. In the co-crystal of protein and DNA, a single strand of DNA fits into the center of the channel formed by the hexamer ring, as depicted in Figure below.
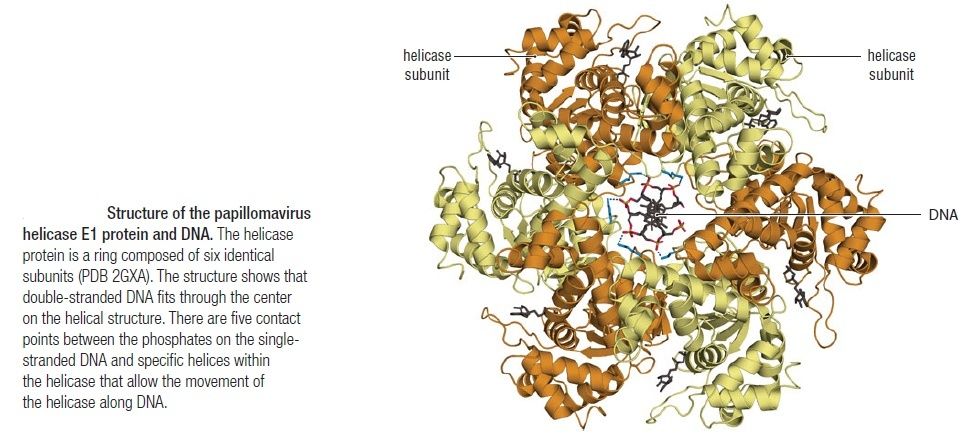
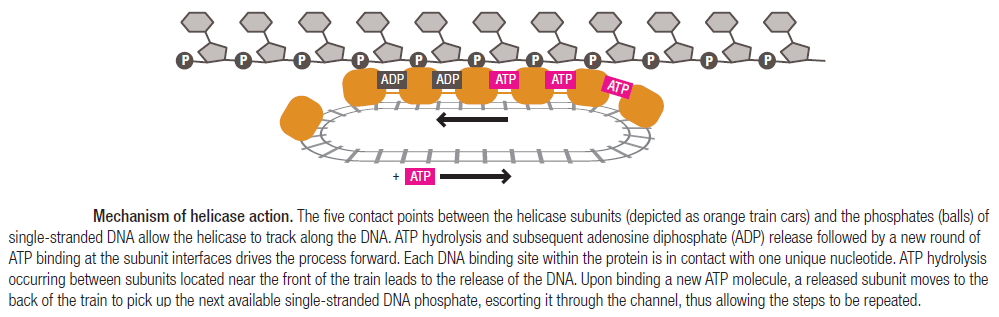
The other strand of the DNA is displaced by the helicase and bound by single-stranded binding proteins. Each monomer possesses a series of DNAbinding loops such that, when the six monomers come together to form the hexamer, the DNA binding loops form a spiral staircase on the inside of the channel which binds the DNA, and move along it one nucleotide at a time. Each movement of the DNA binding loop within each monomer requires adenosine triphosphate (ATP) hydrolysis; thus to move six nucleotides requires six ATP molecules, making DNA unwinding an energy costly process.
In E. coli, the helicase associated with DNA replication is known as DnaB and is a hexamer of six identical subunits. In eukaryotes and archaea, the replicative helicase is called MCM and is a complex of six different proteins, Mcm2–7, which assemble to form a ring. The eukaryotic replicative helicase is conserved throughout evolution and requires accessory factors for helicase activity.

Functions of DnaB 2
This is a fascinating video which shows how DnaB helicase functions:
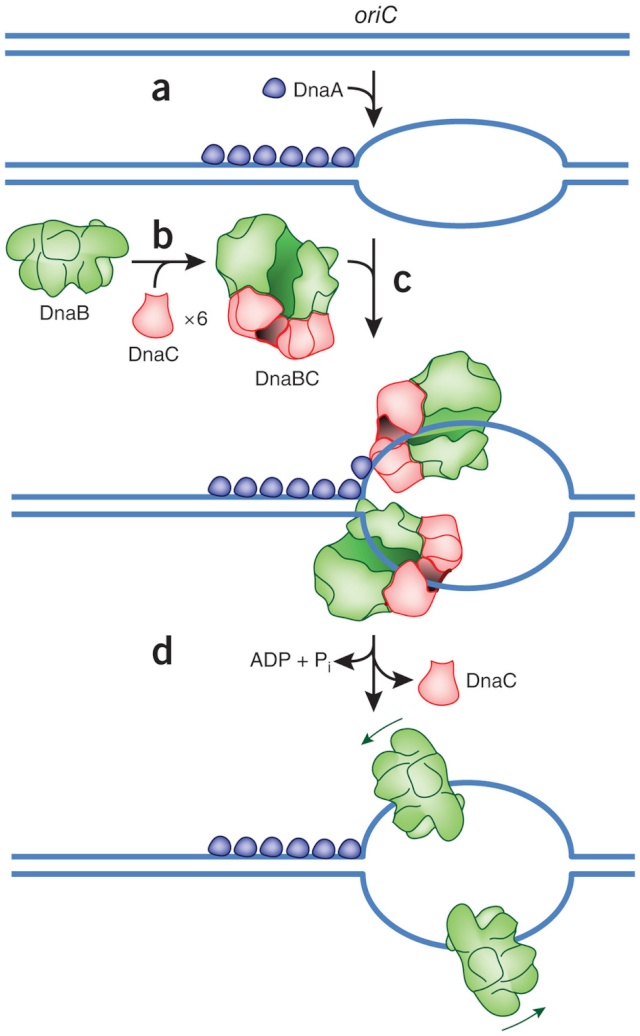
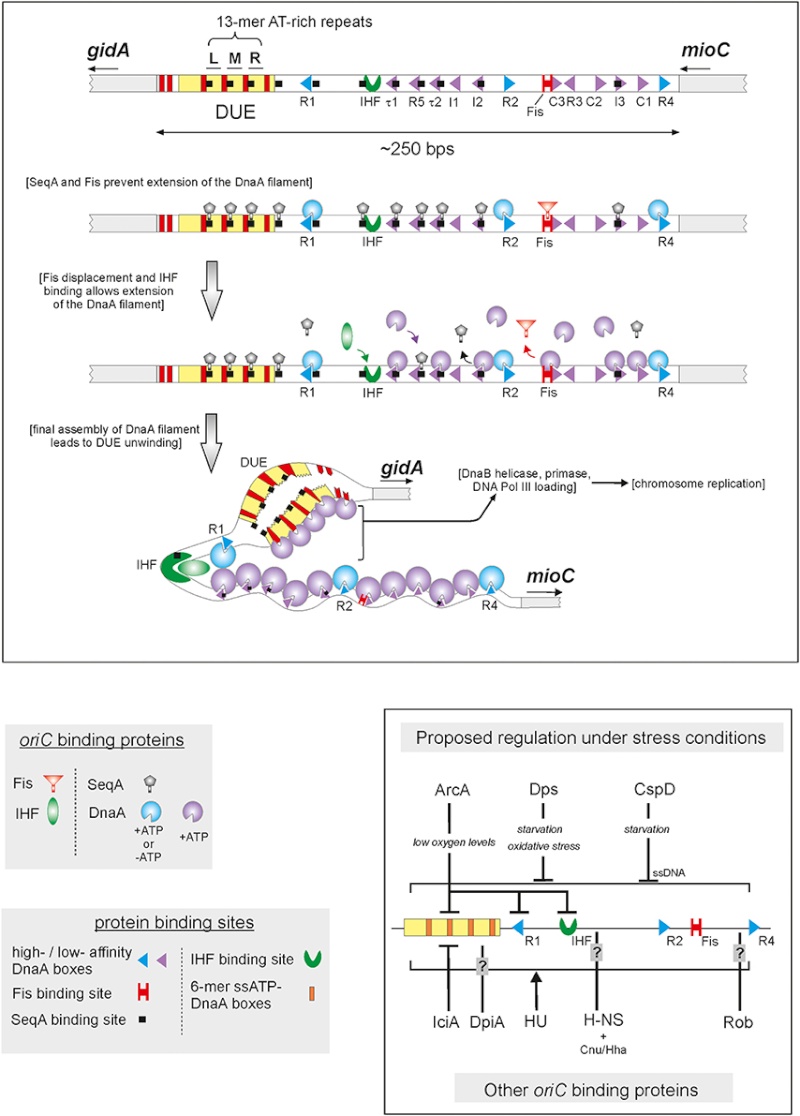
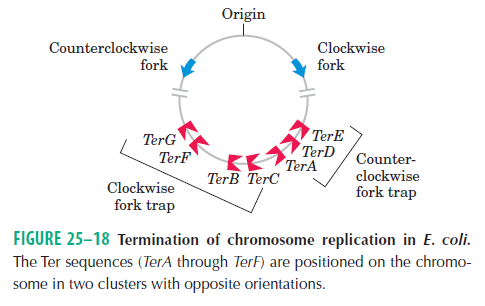
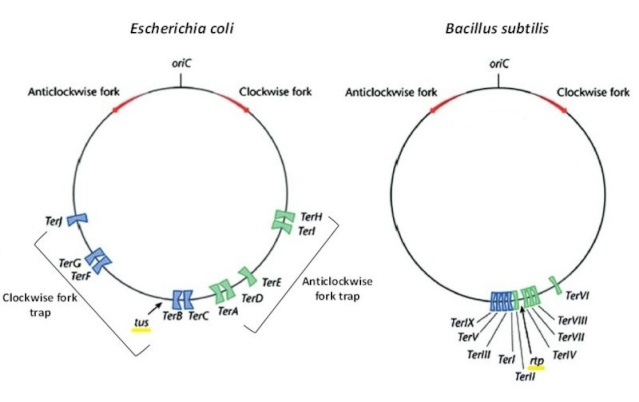
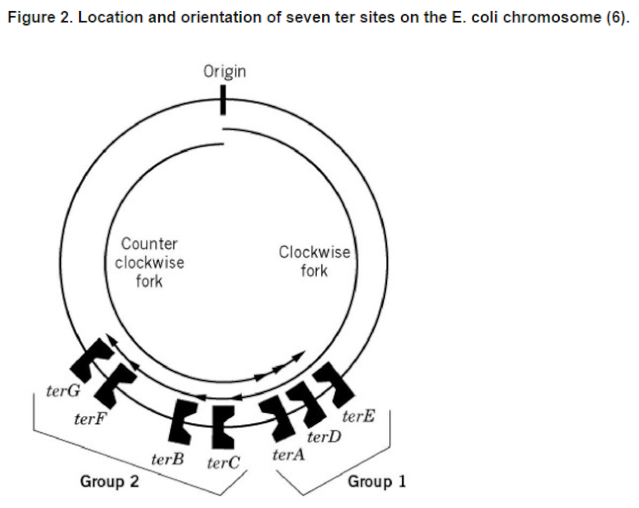
DNA replication begins at the oriC. This is the site where proper replication occurs. The site is 245 base pairs long. This origin contains 4 nine-mers which are the binding sites for a protein called Dna A. The Dna A protein controls the binding of Dna B to the origin. This binding occurs when Dna A initiates the melting of 3 13-mers on the other end of the origin site. This melting of the base pairs causes the Dna B to bind to the origin. Although there is still a problem with Dna B finding the site. So Dna C binds to Dna B to help deliver the complex to the open complex or DNA melting region of origin. After Dna B binds to the 13-mer melted region, this stimulates the binding of Dna G (primase). Thus completing the primosome. The Tao subunit of the DNA polymerase stabilizes and stimulates the Dna B helicase. As the Dna B is stabilized, the protein begins to unwind the dsDNA as it hydrolyzes ATP. The hydrolyzed ATP causes a slight structure change which helps in the ability to connect with the correct sites and move the ssDNA in one direction. Once replication has started there are two main functions the Dna B helicase serves
1. Consistently needs to bind for priming on the lagging strand because the lagging strand produces Okazaki fragments
2. Dna B unwinds the parental DNA to give the pol III templates for the leading and lagging strand, this occurs in the direction of 5’ to 3’. This is the same direction as the replication fork is moving
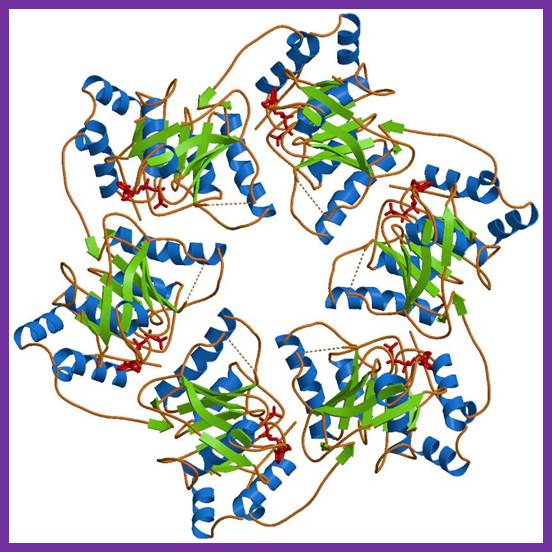
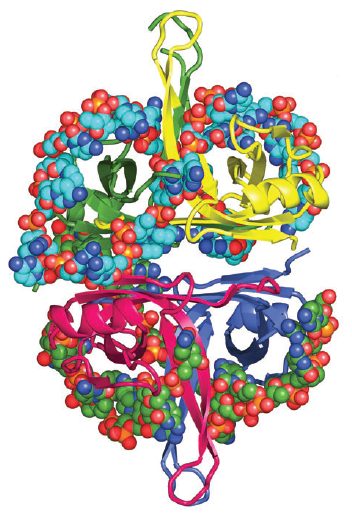
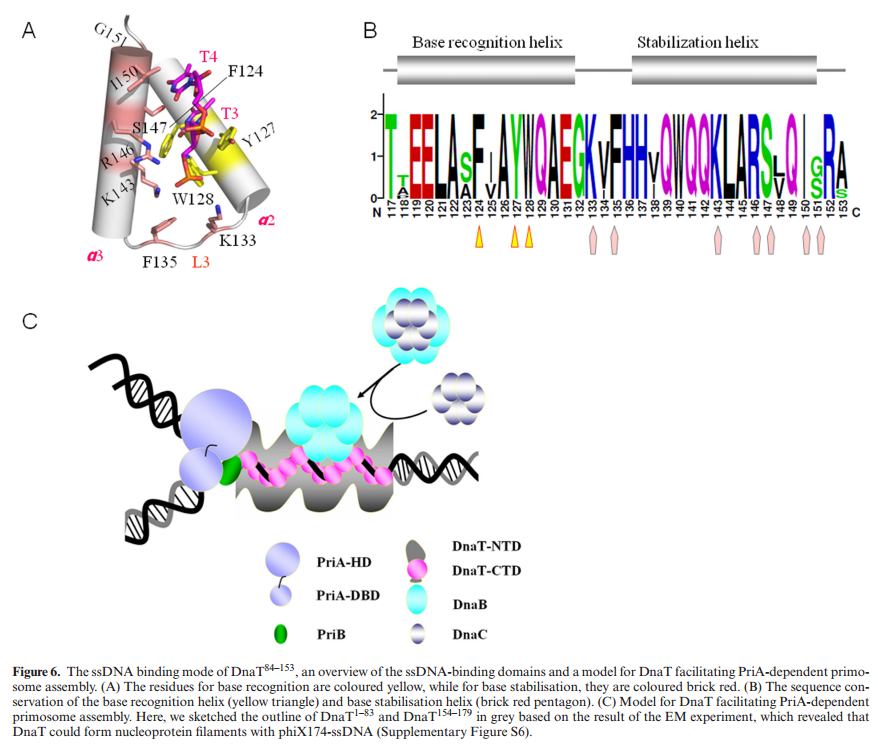
Structure of Hexameric DnaB Helicase and Its Complex with a Domain of DnaG Primase 1
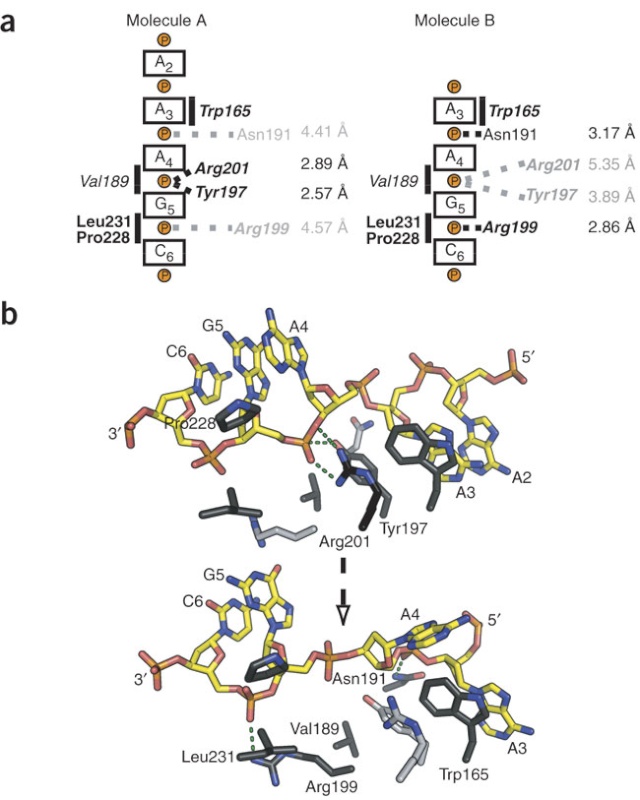
The complex between the DnaB helicase and the DnaG primase unwinds duplex DNA at the eubacterial replication fork and synthesizes the Okazaki RNA primers. The crystal structures of hexameric DnaB and its complex with the helicase binding domain (HBD) of DnaG reveal that within the hexamer the two domains of DnaB pack with strikingly different symmetries to form a distinct two-layered ring structure. Each of three bound HBDs stabilizes the DnaB hexamer in a conformation that may increase its processivity. Three positive, conserved electrostatic patches on the N-terminal domain of DnaB may also serve as a binding site for DNA and thereby guide the DNA to a DnaG active site.

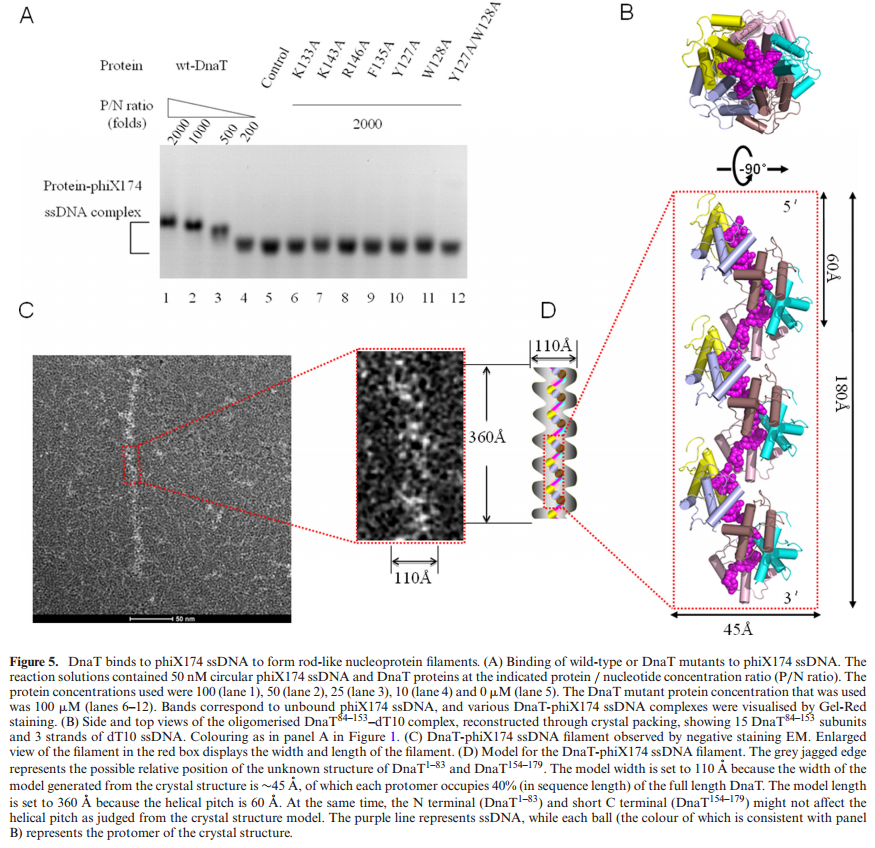
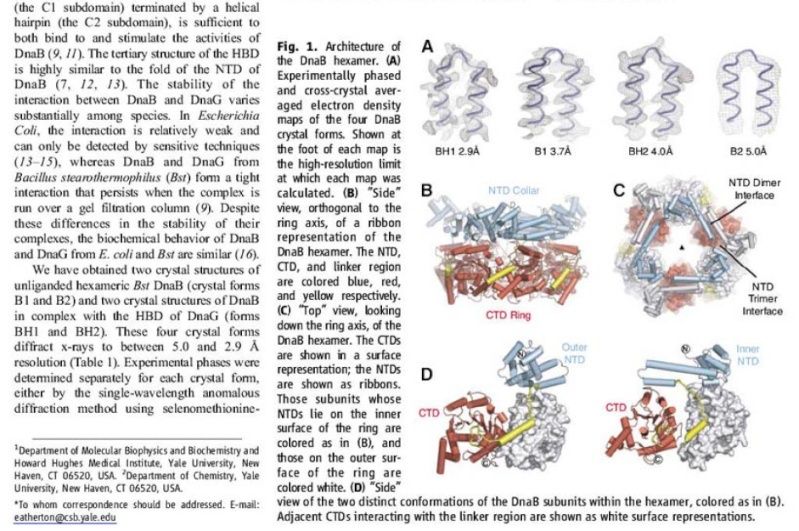
Architecture of the DnaB hexamer.
(A) Experimentally phased and cross-crystal averaged electron density maps of the four DnaB crystal forms. Shown at the foot of each map is the high-resolution limit at which each map was calculated.
(B) “Side” view, orthogonal to the ring axis, of a ribbon representation of the DnaB hexamer. The NTD, CTD, and linker region are colored blue, red, and yellow respectively.
(C) “Top” view, looking down the ring axis, of the DnaB hexamer. The CTDs are shown in a surface representation; the NTDs are shown as ribbons. Those subunits whose NTDs lie on the inner surface of the ring are colored as in (B), and those on the outer surface of the ring are colored white.
(D) “Side” view of the two distinct conformations of the DnaB subunits within the hexamer, colored as in (B). Adjacent CTDs interacting with the linker region are shown as white surface representations.
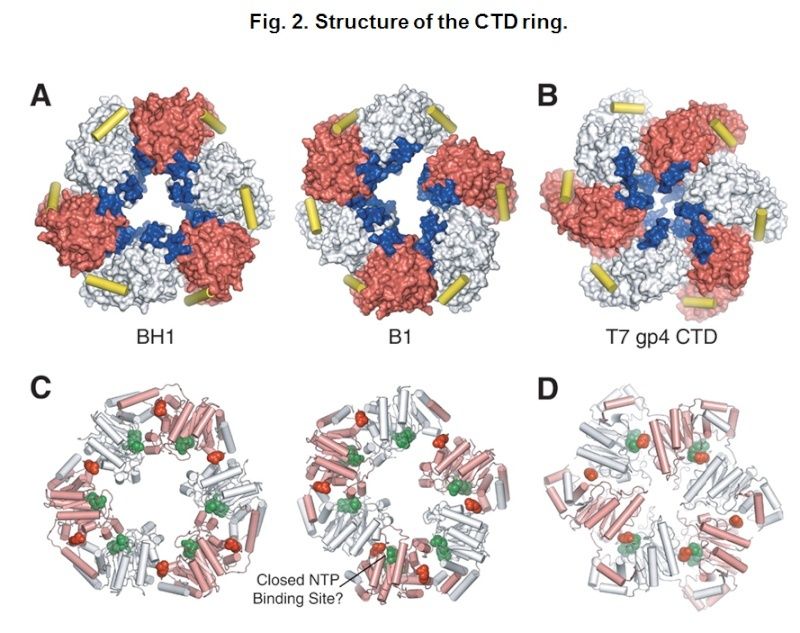
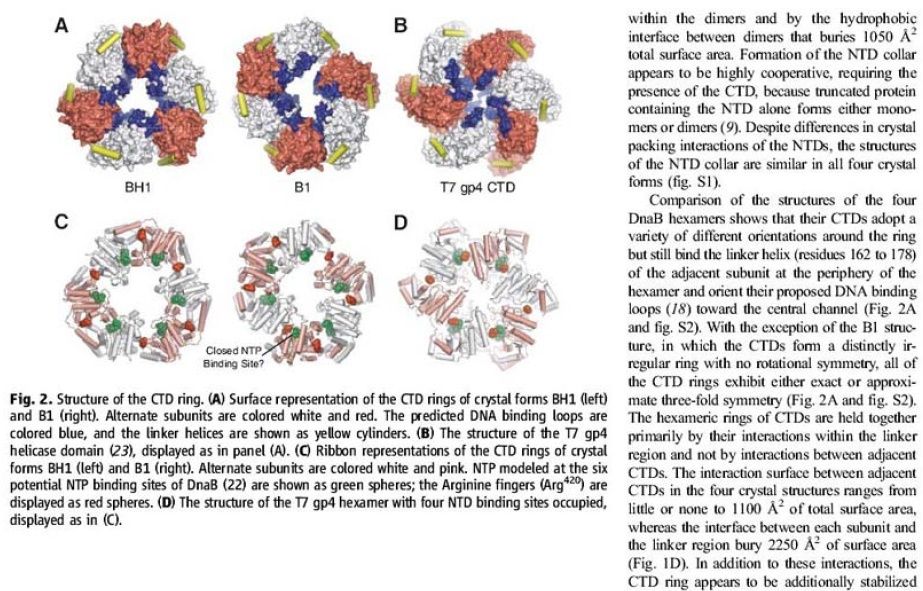
Structure of the CTD ring.
(A) Surface representation of the CTD rings of crystal forms BH1 (left) and B1 (right). Alternate subunits are colored white and red. The predicted DNA binding loops are colored blue, and the linker helices are shown as yellow cylinders.
(B) The structure of the T7 gp4 helicase domain (23), displayed as in panel (A).
(C) Ribbon representations of the CTD rings of crystal forms BH1 (left) and B1 (right). Alternate subunits are colored white and pink. NTP modeled at the six potential NTP binding sites of DnaB (22) are shown as green spheres; the Arginine fingers (Arg420) are displayed as red spheres.
(D) The structure of the T7 gp4 hexamer with four NTD binding sites occupied, displayed as in (C).

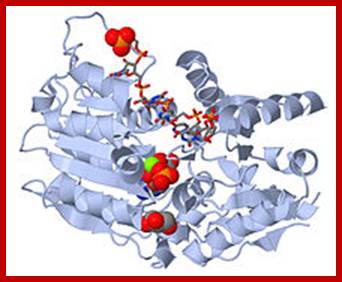
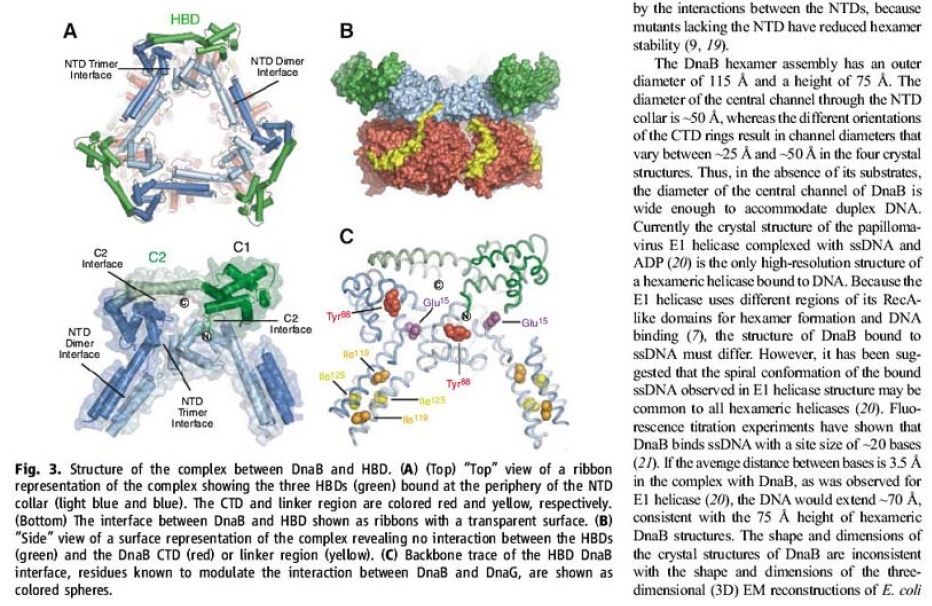
Structure of the complex between DnaB and HBD.
(A) (Top) “Top” view of a ribbon representation of the complex showing the three HBDs (green) bound at the periphery of the NTD collar (light blue and blue). The CTD and linker region are colored red and yellow, respectively. (Bottom) The interface between DnaB and HBD shown as ribbons with a transparent surface.
(B) “Side” view of a surface representation of the complex revealing no interaction between the HBDs (green) and the DnaB CTD (red) or linker region (yellow). [/ltr]
(C) Backbone trace of the HBD DnaB interface, residues known to modulate the interaction between DnaB and DnaG, are shown as colored spheres.

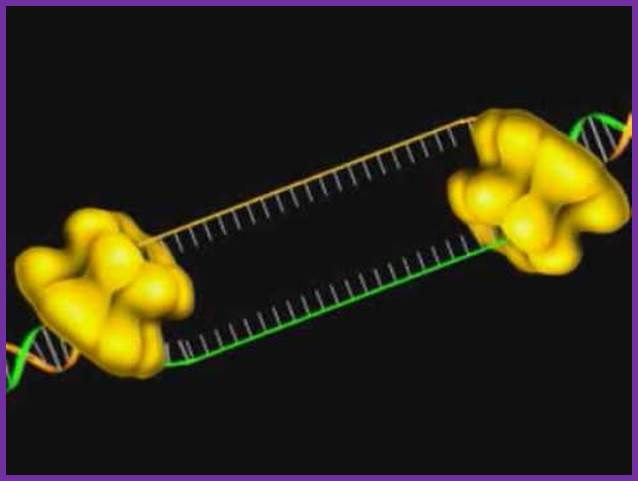
DNA interactions.
(A) (Left) “Top” view of a surface representation of the NTD collar colored blue for positive and red for negative electrostatic potentials. An asterisk highlights the proposed ssDNA binding sites. (Right) A detailed “side” view of the proposed ssDNA binding site boxed in (A).
(B) Speculative model of DnaB complexed with DnaG and replication fork DNA. The proteins are shown in a surface representation (DnaB NTD, light blue; DnaB CTD, red; DnaG HBD, green; DnaG RPD, pink; and DnaG ZBD, orange). The modeled DNA is shown as white- and wheat-colored spheres; the RNA primer is shown in dark blue.






1) http://www.sciencemag.org/content/318/5849/459.abstract
2) http://www.biochem.umd.edu/biochem/kahn/molmachines/replication/dna_b_protein.htm
DNA HELICASES: UNWINDING OF THE DOUBLE HELIX
DNA helicase unwinds double-stranded DNA for copying
Having now considered the key enzymes involved in the actual synthesis of DNA during replication, let us now turn to an enzyme that has an important function before synthesis can actually occur – the unwinding of the double-stranded helix of the parent DNA. When base-paired in the double-stranded helical DNA molecule, nucleotides are inaccessible to polymerases, making copying impossible. Both primase and polymerases need access to single-stranded DNA. To achieve this, the internal base-pairing must be broken and the helix unwound. The first step in this unwinding process is the initial opening of the helix, a step performed by the initiator protein at the origin of replication. Once opened, the unwinding of the double helix to expose single-stranded DNA for copying can begin. This unwinding process is catalyzed by an enzyme called DNA helicase. The cell also needs to open the helix for DNA repair and recombination, and there are a unique set of DNA helicases for this particular case of DNA unwinding. Helicases open the double-stranded DNA and then travel with the replication fork, continuously unwinding the DNA to provide a template for the polymerase to copy. The helicases involved in replication in both bacteria and eukaryotes are composed of six subunits that form a ring structure that surrounds one strand of the DNA. The structure of the replicative DNA helicase from papillomavirusprovides insights into the mechanism of helicase action. This viral helicase is a hexamer of one protein, E1. In the co-crystal of protein and DNA, a single strand of DNA fits into the center of the channel formed by the hexamer ring, as depicted in Figure below.
The other strand of the DNA is displaced by the helicase and bound by single-stranded binding proteins. Each monomer possesses a series of DNAbinding loops such that, when the six monomers come together to form the hexamer, the DNA binding loops form a spiral staircase on the inside of the channel which binds the DNA, and move along it one nucleotide at a time. Each movement of the DNA binding loop within each monomer requires adenosine triphosphate (ATP) hydrolysis; thus to move six nucleotides requires six ATP molecules, making DNA unwinding an energy costly process.
In E. coli, the helicase associated with DNA replication is known as DnaB and is a hexamer of six identical subunits. In eukaryotes and archaea, the replicative helicase is called MCM and is a complex of six different proteins, Mcm2–7, which assemble to form a ring. The eukaryotic replicative helicase is conserved throughout evolution and requires accessory factors for helicase activity.
Functions of DnaB 2
This is a fascinating video which shows how DnaB helicase functions:
DNA replication begins at the oriC. This is the site where proper replication occurs. The site is 245 base pairs long. This origin contains 4 nine-mers which are the binding sites for a protein called Dna A. The Dna A protein controls the binding of Dna B to the origin. This binding occurs when Dna A initiates the melting of 3 13-mers on the other end of the origin site. This melting of the base pairs causes the Dna B to bind to the origin. Although there is still a problem with Dna B finding the site. So Dna C binds to Dna B to help deliver the complex to the open complex or DNA melting region of origin. After Dna B binds to the 13-mer melted region, this stimulates the binding of Dna G (primase). Thus completing the primosome. The Tao subunit of the DNA polymerase stabilizes and stimulates the Dna B helicase. As the Dna B is stabilized, the protein begins to unwind the dsDNA as it hydrolyzes ATP. The hydrolyzed ATP causes a slight structure change which helps in the ability to connect with the correct sites and move the ssDNA in one direction. Once replication has started there are two main functions the Dna B helicase serves
1. Consistently needs to bind for priming on the lagging strand because the lagging strand produces Okazaki fragments
2. Dna B unwinds the parental DNA to give the pol III templates for the leading and lagging strand, this occurs in the direction of 5’ to 3’. This is the same direction as the replication fork is moving
Structure of Hexameric DnaB Helicase and Its Complex with a Domain of DnaG Primase 1
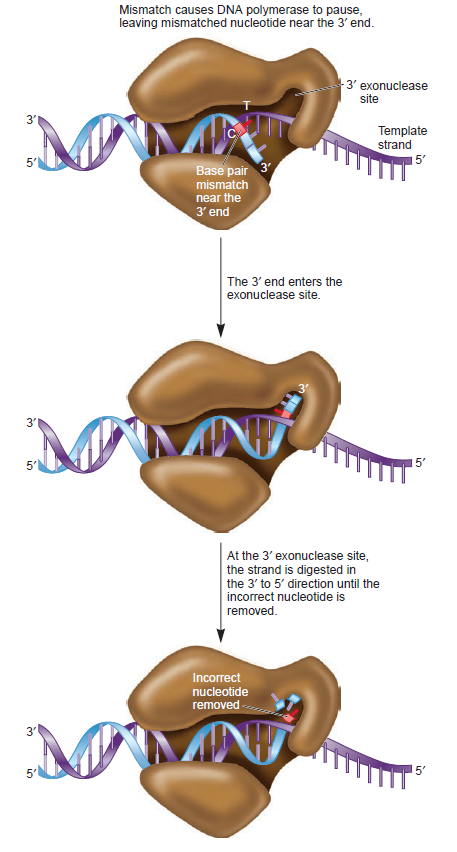
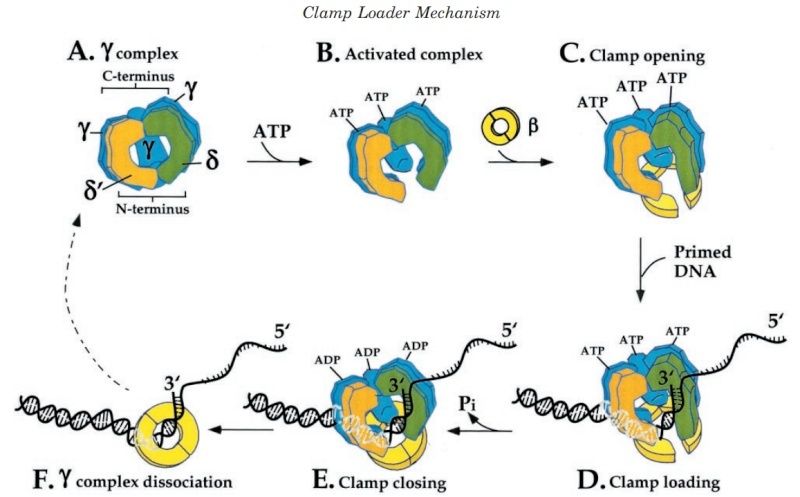
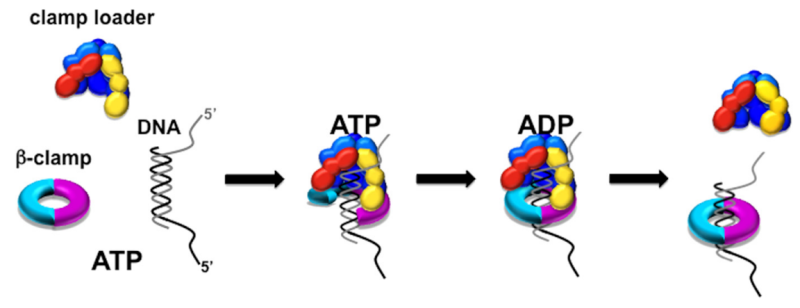
The complex between the DnaB helicase and the DnaG primase unwinds duplex DNA at the eubacterial replication fork and synthesizes the Okazaki RNA primers. The crystal structures of hexameric DnaB and its complex with the helicase binding domain (HBD) of DnaG reveal that within the hexamer the two domains of DnaB pack with strikingly different symmetries to form a distinct two-layered ring structure. Each of three bound HBDs stabilizes the DnaB hexamer in a conformation that may increase its processivity. Three positive, conserved electrostatic patches on the N-terminal domain of DnaB may also serve as a binding site for DNA and thereby guide the DNA to a DnaG active site.
Architecture of the DnaB hexamer.
(A) Experimentally phased and cross-crystal averaged electron density maps of the four DnaB crystal forms. Shown at the foot of each map is the high-resolution limit at which each map was calculated.
(B) “Side” view, orthogonal to the ring axis, of a ribbon representation of the DnaB hexamer. The NTD, CTD, and linker region are colored blue, red, and yellow respectively.
(C) “Top” view, looking down the ring axis, of the DnaB hexamer. The CTDs are shown in a surface representation; the NTDs are shown as ribbons. Those subunits whose NTDs lie on the inner surface of the ring are colored as in (B), and those on the outer surface of the ring are colored white.
(D) “Side” view of the two distinct conformations of the DnaB subunits within the hexamer, colored as in (B). Adjacent CTDs interacting with the linker region are shown as white surface representations.
Structure of the CTD ring.
(A) Surface representation of the CTD rings of crystal forms BH1 (left) and B1 (right). Alternate subunits are colored white and red. The predicted DNA binding loops are colored blue, and the linker helices are shown as yellow cylinders.
(B) The structure of the T7 gp4 helicase domain (23), displayed as in panel (A).
(C) Ribbon representations of the CTD rings of crystal forms BH1 (left) and B1 (right). Alternate subunits are colored white and pink. NTP modeled at the six potential NTP binding sites of DnaB (22) are shown as green spheres; the Arginine fingers (Arg420) are displayed as red spheres.
(D) The structure of the T7 gp4 hexamer with four NTD binding sites occupied, displayed as in (C).
Structure of the complex between DnaB and HBD.
(A) (Top) “Top” view of a ribbon representation of the complex showing the three HBDs (green) bound at the periphery of the NTD collar (light blue and blue). The CTD and linker region are colored red and yellow, respectively. (Bottom) The interface between DnaB and HBD shown as ribbons with a transparent surface.
(B) “Side” view of a surface representation of the complex revealing no interaction between the HBDs (green) and the DnaB CTD (red) or linker region (yellow). [/ltr]
(C) Backbone trace of the HBD DnaB interface, residues known to modulate the interaction between DnaB and DnaG, are shown as colored spheres.
DNA interactions.
(A) (Left) “Top” view of a surface representation of the NTD collar colored blue for positive and red for negative electrostatic potentials. An asterisk highlights the proposed ssDNA binding sites. (Right) A detailed “side” view of the proposed ssDNA binding site boxed in (A).
(B) Speculative model of DnaB complexed with DnaG and replication fork DNA. The proteins are shown in a surface representation (DnaB NTD, light blue; DnaB CTD, red; DnaG HBD, green; DnaG RPD, pink; and DnaG ZBD, orange). The modeled DNA is shown as white- and wheat-colored spheres; the RNA primer is shown in dark blue.
1) http://www.sciencemag.org/content/318/5849/459.abstract
2) http://www.biochem.umd.edu/biochem/kahn/molmachines/replication/dna_b_protein.htm
Last edited by Admin on Wed Nov 25, 2015 8:51 am; edited 8 times in total