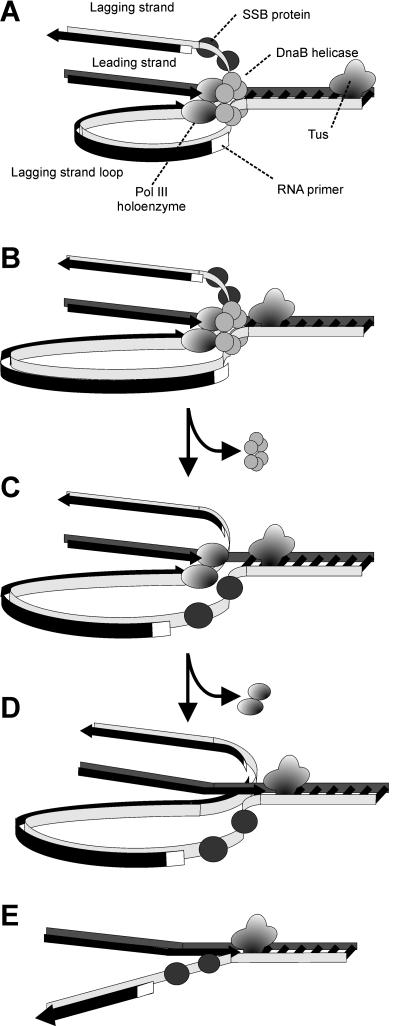
DNA damage and repairFate of the replisome following arrest by UV-induced DNA damage in Escherichia coli. 7
ABSTRACT
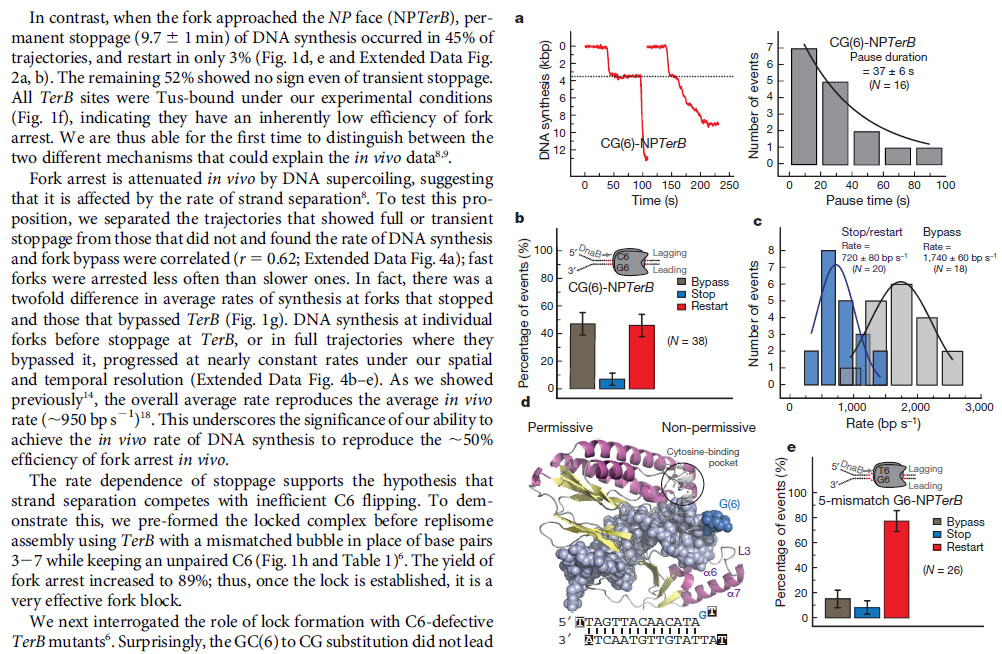
Accurate replication in the presence of DNA damage is essential to genome stability and viability in all cells. In Escherichia coli, DNA replication forks blocked by UV-induced damage undergo a partial resection and RecF-catalyzed regression before synthesis resumes. These processing events generate distinct structural intermediates on the DNA. However, the fate and behavior of the stalled replisome remains a central uncharacterized question. Here, we use thermosensitive mutants to show that the replisome’s polymerases uncouple and transiently dissociate from the DNA in vivo. Inactivation of α, β, or τ subunits within the replisome is sufficient to signal and induce the RecF-mediated processing events observed following UV damage. By contrast, the helicase–primase complex (DnaB and DnaG) remains critically associated with the fork, leading to a loss of fork integrity, degradation, and aberrant intermediates when disrupted. The results reveal a dynamic replisome, capable of partial disassembly to allow access to the obstruction, while retaining subunits that maintain fork licensing and direct reassembly to the appropriate location after processing has occurred.The replisome consists of several, multisubunit protein complexes and is responsible for duplicating the genome. In Escherichia coli, it is comprised of three DNA polymerase complexes tethered to the DNA template by dimeric processivity factors, a τ complex that couples leading and lagging strand synthesis, and a helicase–primase complex that separates the duplex DNA and primes lagging strand synthesis (1–3).When the replisome encounters DNA damage that blocks its progression, the potential for mutagenesis, rearrangements, and lethality increases significantly. Replication in the presence of DNA damage can generate mutations if the wrong base is incorporated, rearrangements if it resumes from the wrong site, or lethality if the obstructing lesion cannot be overcome. Following the arrest of replication at UV-induced damage, the nascent lagging strand is partially resected by the combined action of the RecQ helicase In prokaryotes RecQ is necessary for plasmid recombination and DNA repair from UV-light, free radicals, and alkylating agents. This protein can also reverse damage from replication errors. 8 The RecQ family of helicases are enzymes that unwind DNA so that replication, transcription, and DNA repair can occur. 9RecQ helicases are highly conserved from bacteria to man. 10, 11. Germline mutations in three of the five known family members in humans give rise to debilitating disorders that are characterized by, amongst other things, a predisposition to the development of cancer. One of these disorders — Bloom's syndrome — is uniquely associated with a predisposition to cancers of all types. So how do RecQ helicases protect against cancer? They seem to maintain genomic stability by functioning at the interface between DNA replication and DNA repair. RecJ nuclease (4, 5). The RecJ exonuclease from Escherichia coli degrades single-stranded DNA (ssDNA) in the 5′–3′ direction and participates in homologous recombination and mismatch repair. 12RecF-O-R, along with RecA, limit this degradation and promote a transient regression of the DNA branch point, which is thought to be important for restoring the damaged region to a form that can be acted on by repair enzymes or translesion DNA polymerases (4–10). These processing events generate distinct structural intermediates on the DNA, a technique that allows one to identify the shape and structure of DNA molecules (5, 11).Although the processing that occurs on the DNA is well characterized, little is known about the behavior or composition of the replisome itself during these events. If the replisome remains bound to the arresting lesion, it may sterically obstruct repair or bypass from occurring. Conversely, complete dissociation of the replisome would likely abolish the licensing for the replication fork and expose DNA ends that have the potential to recombine, generating deletions, duplications, or rearrangements on the chromosome. Recent studies in vitro have suggested that dynamic interactions between replisome components may play a role in allowing the machinery to overcome specific challenges such as collisions with the transcription apparatus or DNA-bound proteins (1, 12, 13). UV-induced photoproducts is a biologically relevant lesion that is known to block the progression of the replisome when located in the leading strand template (6,14–16). The results demonstrate that the DNA polymerases can dissociate from the replisome in a modular manner without compromising the integrity of the replication fork. Dissociation of the DNA polymerase from the replisome is sufficient and can serve to initiate the processing of the replication fork DNA via the RecF pathway, similar to that seen when replication is arrested by UV-induced damage. By comparison, the helicase complex remains associated with the replication fork throughout the recovery process. If the helicase is disrupted, aberrant intermediates, degradation, and loss of fork integrity ensue. We propose that the retention of the helicase is needed to maintain licensing for the replication fork and direct reassembly to the appropriate location after processing has occurred.A schematic of each of the components of the replisome tested in this study and their function is presented in Fig. 1A. Temperature-sensitive mutants exist in subunits from each of replisome’s complexes for which viability or functionality is supported at 30 °C, but not at 42 °C (Fig. 1B). Although replication proceeds normally at the permissive temperature, it rapidly decreases following inactivation of the thermosensitive protein at the restrictive temperature, similar to that seen after UV irradiation (Fig. 1C). The exception to this is in the proofreading subunit ε, encoded by dnaQts, which is mutagenic at the restrictive temperature, but is not essential for viability or replication (17).
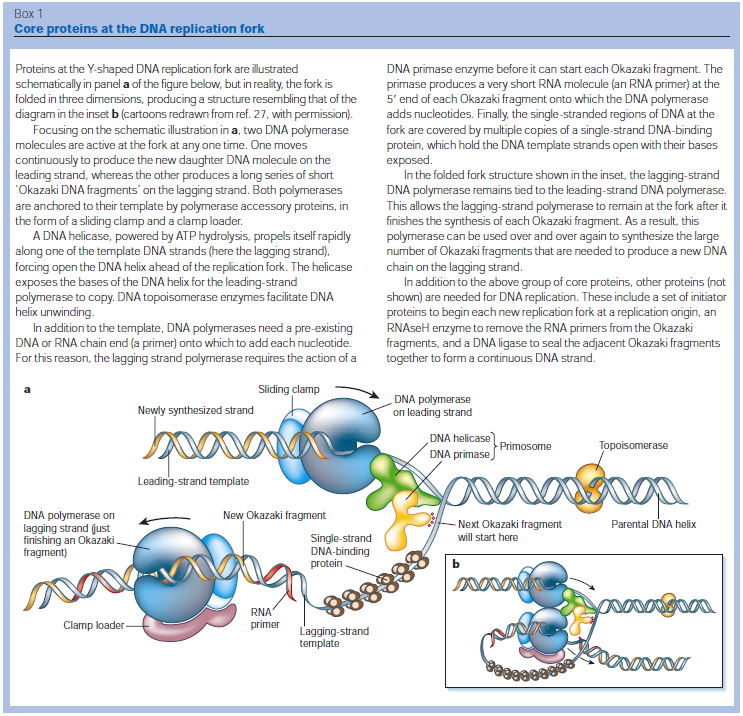
 Replication is disrupted by UV-induced damage or following inactivation of the DNA polymerase, τ complex, or helicase–primase complex. (A) A diagram of the replisome, indicating the subunits of each protein complex. (B) Thermosensitive mutants that inactivate the polymerase core, τ complex, or helicase complex are viable at 30 °C but fail to grow at the restrictive temperature of 42 °C following overnight incubation. (C) The rate of DNA synthesis is inhibited following UV-induced damage or inactivation of the replisome’s essential subunits. Wild-type or mutant cultures, grown at 30 °C were pulse-labeled with 1 µCi per 10 µg/mL [3H]thymidine for 2 min at the indicated times following mock treatment (open symbols), 50 J/m2 UV irradiation (filled symbols), or a shift to 42 °C (filled symbols). The amount of radioactivity incorporated into the DNA, relative to pretreated cultures is plotted. Error bars represent SE of two experiments.
Replication is disrupted by UV-induced damage or following inactivation of the DNA polymerase, τ complex, or helicase–primase complex. (A) A diagram of the replisome, indicating the subunits of each protein complex. (B) Thermosensitive mutants that inactivate the polymerase core, τ complex, or helicase complex are viable at 30 °C but fail to grow at the restrictive temperature of 42 °C following overnight incubation. (C) The rate of DNA synthesis is inhibited following UV-induced damage or inactivation of the replisome’s essential subunits. Wild-type or mutant cultures, grown at 30 °C were pulse-labeled with 1 µCi per 10 µg/mL [3H]thymidine for 2 min at the indicated times following mock treatment (open symbols), 50 J/m2 UV irradiation (filled symbols), or a shift to 42 °C (filled symbols). The amount of radioactivity incorporated into the DNA, relative to pretreated cultures is plotted. Error bars represent SE of two experiments.
 Model of replisome at UV-induced damage. Upon encountering an arresting lesion (PD, pyrimidine dimer) (i), DNA synthesis becomes uncoupled and the polymerases transiently dissociate. (ii) This serves as a signal to initiate the replication fork DNA processing by the RecF-pathway gene products (gray circles) allowing repair enzymes (NER) or translesion polymerases to access the lesion. (iii) The helicase–primase complex remains bound to the template DNA and serves to maintain the licensing and integrity of the replication fork, directing replisome reassembly to the correct location once the lesion has been processed.Cellular Characterization of the Primosome and Rep Helicase in Processing and Restoration of Replication following Arrest by UV-Induced DNA Damage in Escherichia coli
Model of replisome at UV-induced damage. Upon encountering an arresting lesion (PD, pyrimidine dimer) (i), DNA synthesis becomes uncoupled and the polymerases transiently dissociate. (ii) This serves as a signal to initiate the replication fork DNA processing by the RecF-pathway gene products (gray circles) allowing repair enzymes (NER) or translesion polymerases to access the lesion. (iii) The helicase–primase complex remains bound to the template DNA and serves to maintain the licensing and integrity of the replication fork, directing replisome reassembly to the correct location once the lesion has been processed.Cellular Characterization of the Primosome and Rep Helicase in Processing and Restoration of Replication following Arrest by UV-Induced DNA Damage in Escherichia coli 4
Following arrest by UV-induced DNA damage, replication is restored through a sequence of steps that involve partial resection of the nascent DNA by RecJ and RecQ, branch migration and processing of the fork DNA surrounding the lesion by RecA and RecF-O-R, and resumption of DNA synthesis once the blocking lesion has been repaired or bypassed. In vitro, the primosomal proteins (
PriA, PriB, and PriC) and
Rep are capable of initiating replication from synthetic DNA fork structures, and they have been proposed to catalyze these events when replication is disrupted by certain impediments in vivo. Here, we characterized the role that PriA, PriB, PriC, and Rep have in processing and restoring replication forks following arrest by UV-induced DNA damage. We show that the partial degradation and processing of the arrested replication fork occurs normally in both rep and primosome mutants. In each mutant, the nascent degradation ceases and DNA synthesis initially resumes in a timely manner, but the recovery then stalls in the absence of PriA, PriB, or Rep.
The results demonstrate a role for the primosome and Rep helicase in overcoming replication forks arrested by UV-induced damage in vivo and suggest that these proteins are required for the stability and efficiency of the replisome when DNA synthesis resumes but not to initiate de novo replication downstream of the lesion.PriA, PriB, and PriC were originally identified as proteins required for replication of single-strand ϕX174 phage DNA in vitro and in vivo (70, 71). In vitro, the proteins function as a complex that is required for processive priming to occur behind the replicative helicase, DnaB (1, 2). PriA initially binds a hairpin structure on the ϕX174 chromosome, followed by PriB, DnaT, and PriC. The resulting complex then recruits DnaC, which loads the DnaB helicase onto the chromosome. The DnaG primase is then able to associate with DnaB to synthesize RNA primers. While DnaG and DnaB are sufficient for primer synthesis on ϕX174 DNA (1), specific and processive priming of single-stranded DNA binding protein-coated phage DNA requires PriA (2). In vivo, conversion of ϕX174 from its plus-strand form to its minus-strand replication intermediate requires PriA and other Escherichia coli host proteins (40). E. coli strains lacking PriA have reduced viability, growth rates, and culture densities relative to wild-type cells (36). priA mutants are also constitutively induced for the SOS response, and cells lacking PriA produce filaments extensively (49). Taken together, these observations led early researchers to propose that the primosomal proteins promote efficient priming for Okazaki fragments during lagging-strand replication (35, 38).Replication forks must deal with a variety of obstacles that may impede their progress, including DNA-bound proteins, secondary structures, strand breaks, and adducts or damage to the DNA bases themselves. With respect to DNA base damage, UV irradiation with 254-nm light has often served as a model to address the question of how replication recovers following encounters with this form of impediment. UV irradiation induces two primary photoproducts, cis, syn-cyclobutane pyrimidine dimers (CPDs) and 6,4 pyrimidine-pyrimidone photoproducts (6-4 PPs) (59, 67, 68). Although these lesions block DNA polymerases and arrest replication (28, 58), growing E. coli cultures survive doses that produce more than 2,000 lesions per genome (30), indicating that cells contain efficient mechanisms to process these lesions when they are encountered during replication.The recovery of replication following arrest by UV-induced DNA damage occurs through a sequence of well-characterized steps. Following arrest, the nascent lagging strand is partially degraded by the combined action of the RecJ nuclease and RecQ helicase. This processing is thought to restore the lesion-containing region to a double-stranded form that can be accessed and repaired by the nucleotide excision repair complex (17). RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli 5
All cells must faithfully replicate their genomes in order to reproduce. However,
if not repaired, DNA damage that blocks replication can lead to a loss of genomic stability, mutations, or cell death. Despite the importance of the process by which replication recovers, the cellular mechanism(s) by which this occurs in DNA repair proficient cells remains largely uncharacterized. Irradiation of cells with near UV light induces lesions in the DNA which block replication. In E. coli, replication is transiently inhibited following a moderate dose of UV irradiation, but it eficiently recovers following the removal of the UV-induced lesions . The eficient recovery of replication in wild-type cells is accompanied by the partial degradation of the nascent DNA at the replication fork prior to the resumption of DNA synthesis . However, it is not known whether this degradation is required for, or contributes in any way to, the normal recovery process. The resumption of replication following UV-induced DNA damage is largely dependent upon the removal of the lesions by
nucleotide excision repair . However, a large body of work with repair-deficient mutants has shown that UV irradiation can lead to recombination events when replication forks encounter DNA damage that cannot be repaired. In these mutants, the recovery of replication is severely inhibited, resulting in loss of semiconservative replication, high frequencies of chromosomal exchanges, and extensive cell death . In contrast, these recombination events are eficiently suppressed in normal, repair-proficient cells; survival is greatly enhanced and the recovery of replication is much more eficient, suggesting that the normal mechanism of recovery may be quite different from that observed in repair-deficient mutants.
In addition to removal of the lesions, however, the recovery of replication also requires the function of RecA and the recF pathway proteins Historically, because most of these proteins were identified through recombination
RecQ helicase and RecJ nuclease provide complementary functions to resect DNA for homologous recombination 6
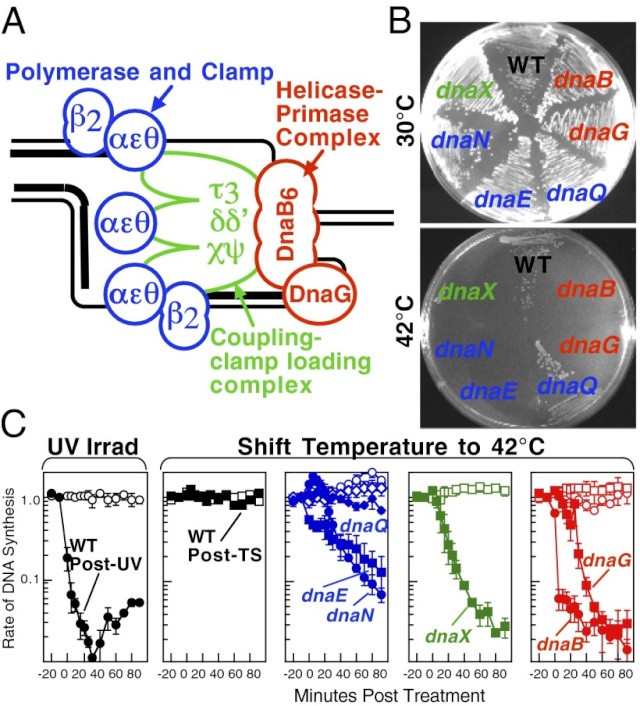
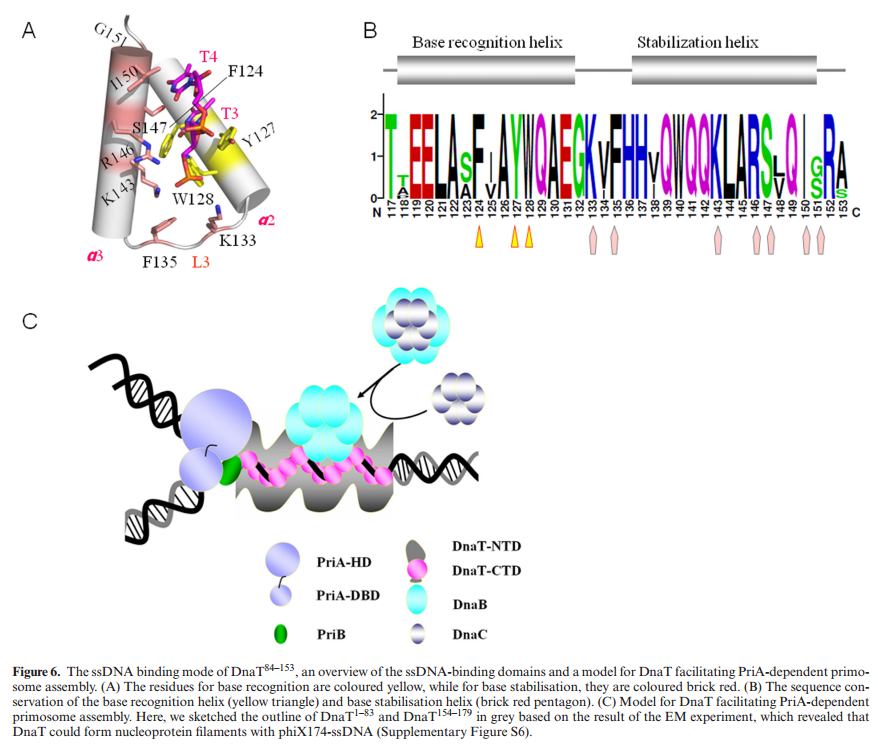
Recombinational DNA repair by the RecF pathway of Escherichia coli requires the coordinated activities of RecA, RecFOR, RecQ, RecJ, single-strand DNA binding (SSB) proteins.These proteins facilitate formation of homologously paired joint molecules between linear double-stranded (dsDNA) and supercoiled DNA. Repair starts with resection of the broken dsDNA by RecQ, a 3′→5′ helicase, RecJ, a 5′→3′ exonuclease, and SSB protein. The ends of a dsDNA break can be blunt-ended, or they may possess either 5′- or 3′-single-stranded DNA (ssDNA) overhangs of undefined length. Here we show that RecJ nuclease alone can initiate nucleolytic resection of DNA with 5′-ssDNA overhangs, and that RecQ helicase can initiate resection of DNA with blunt-ends or 3′-ssDNA overhangs by DNA unwinding. We establish that in addition to its well-known ssDNA exonuclease activity, RecJ can display dsDNA exonuclease activity, degrading 100–200 nucleotides of the strand terminating with a 5′-ssDNA overhang. The dsDNA product, with a 3′-ssDNA overhang, is an optimal substrate for RecQ, which unwinds this intermediate to reveal the complementary DNA strand with a 5′-end that is degraded iteratively by RecJ. On the other hand, RecJ cannot resect duplex DNA that is either blunt-ended or terminated with 3′-ssDNA; however, such DNA is unwound by RecQ to create ssDNA for RecJ exonuclease. RecJ requires interaction with SSB for exonucleolytic degradation of ssDNA but not dsDNA. Thus, complementary action by RecJ and RecQ permits initiation of recombinational repair from all dsDNA ends: 5′-overhangs, blunt, or 3′-overhangs. Such helicase–nuclease coordination is a common mechanism underlying resection in all organisms.Homologous recombination is a relatively error-free mechanism to repair double-stranded DNA (dsDNA) breaks (DSBs) and single-stranded DNA (ssDNA) gaps, which are produced by UV light, γ-irradiation, and chemical mutagens (1). In wild-type Escherichia coli, the labor of recombinational repair is divided between the RecBCD and RecF pathways of recombination, which are responsible for the repair of DSBs and ssDNA gaps, respectively (2–5). However, the proteins of the RecF pathway are capable of DSB repair, as well as ssDNA gap repair: in recBC mutant cells containing the suppressor mutations, sbcB andsbcC (suppressors of recBC), the proteins of the RecF pathways provide the needed recombinational DNA repair functions (2, 6).Consistent with this, in the absence of either repair or nascent DNA degradation, the recovery of replication is delayed, and both survival and recovery become dependent on translesion synthesis by DNA polymerase V (12, 13). RecF, RecO, and RecRlimit the RecJ/RecQ-mediated degradation and enhance the formation of RecA filaments at the arrested region (11, 14, 60, 64). Biochemical characterizations suggest that the RecA filament formed in the presence of RecFOR is capable of promoting branch migration at the fork in a manner that could promote regression away from the lesion and subsequently reset the 3′ end of the fork once the impediment has been removed or overcome (47, 60, 64, 69). In vivo, cells lacking any one of these gene products fail to resume DNA synthesis, and the DNA at the replication fork is extensively degraded (11, 14, 15).Several lines of evidence suggest that Rep and the primosome also participate in restoring replication following arrest at a UV-induced lesion, either through direct resumption of the arrested replisome or de novo initiation of a replisome downstream of the arrest site. Both priA and rep contribute to the DNA synthesis that occurs during recombinational processes (26, 32, 41, 52, 65). Although no single gene by itself is essential for viability, double mutants in priA and priC or priA and rep are lethal, and both priA and rep mutants are hypersensitive to DNA damage (53). It has also been widely postulated that frequent replication disruptions by endogenous DNA damage in vivo account for the poor growth and low viability of priA and rep mutants (8, 45, 57). In addition, one study has reported a delayed recovery of DNA synthesis in PriA mutants following low doses of UV light (51). In vitro, the addition of PriA and PriB, PriA and PriC, or PriC and Rep allows DNA synthesis to occur at synthetic DNA fork structures in the presence of the other core replication proteins (25, 26). However, the role of PriA, PriB, PriC, and Rep in the progressive steps of resection, processing, or resumption following replication arrest at UV-induced DNA damage has not been directly examined in vivo. Here, we characterize the molecular events that occur during the progressive steps associated with the recovery of replication in UV-irradiated cultures of mutants lacking each of these gene products.Two models that have been proposed for how the primosome and Rep helicase participate in restoring an active replisome following arrest by DNA damage are summarized in Fig. 6. Both models propose late functions for the primosome and Rep helicase but differ in the mechanism by which they promote replication recovery. The first model proposes that following arrest, the replisome and helicase are disrupted. Combinations of either PriA, PriB, or Rep with PriC participate in the displacement of the nascent lagging strand. These proteins then facilitate a transient loading of the helicase and primase complex on the leading-strand template, which serves as a primer, allowing a replisome to reinitiate downstream from the site of arrest (Fig. 6A). This model arose from the observation that, in vitro, the helicase activity of either PriA or Rep was capable of displacing the strands of a synthetic replication fork structure. In the presence of the helicase loader, DnaC, this is sufficient for the helicase and primase to prime the resulting single-stranded regions that are generated on the leading- and lagging-strand templates in vitro (22, 27).The second model proposes that the primosome's primary contribution relates to enhancing the replisome's stability or priming efficiency during basal replication. Following arrest by UV-induced DNA damage, the helicase remains associated with the lagging strand, but other components of the holoenzyme may be displaced or disrupted. RecQ and RecJ contribute to the displacement and partial degradation of the nascent lagging strand, while the RecFOR proteins, together with RecA, process the fork DNA such that the lesion can either be repaired or bypassed. Once the block to replication has been overcome, the replisome can resume from the original arrest site. However, reestablishing an efficient replisome requires the primosome protein PriA and, to a lesser extent, PriB and PriC to coordinate the helicase/primase complex with the progressing replisome. The Rep helicase may also contribute to this reaction by helping to clear the region of other protein factors, such as recombination proteins, repair enzymes, or translesion polymerases, that may impair or compete with the replisome's ability to bind its forked substrate (Fig. 6B).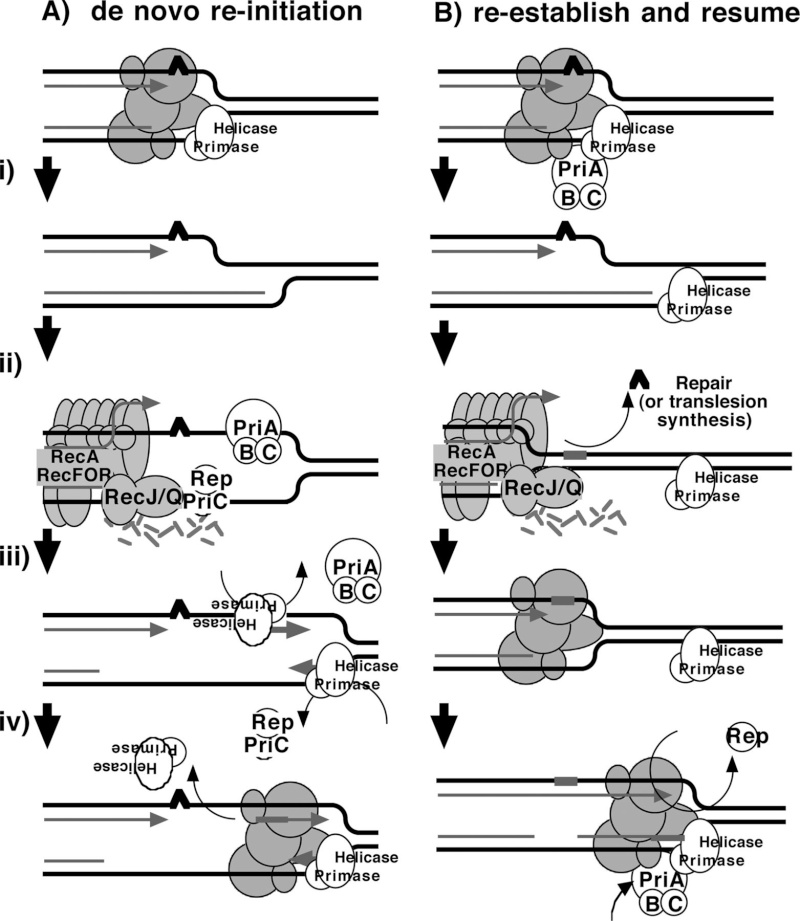 FIG 6Two models for primosome and Rep function following disruption by DNA damage. (A) A model proposing that PriA and Rep function specifically to reinitiate DNA synthesis following disruption events. (i) Following the disruption of the replication machinery (grayed circles) by DNA damage (∧), (ii) PriA or Rep functions in a reaction to transiently load DnaB and DnaG to prime the leading strand and then (iii) stably load DnaB and DnaG on the lagging strand (22, 27). (iv) The leading-strand primer allows for the de novo formation of an active replisome downstream from the site of disruption. (B) A model in which PriA and Rep are required by the replisome to maintain efficient replication. (i) Following disruption by DNA damage, the recovery of DNA synthesis requires that the lesion is either repaired (ii) or bypassed (iii) by translesion synthesis (not shown), as found in previous studies (13). (iv) Since PriA and Rep are needed to maintain replication in the absence of damage, PriA and Rep would also be required for an active replisome to be maintained once the replisome is reestablished and DNA synthesis resumes.Structural Insight into the DNA-Binding Mode of the Primosomal Proteins PriA, PriB, and DnaT
FIG 6Two models for primosome and Rep function following disruption by DNA damage. (A) A model proposing that PriA and Rep function specifically to reinitiate DNA synthesis following disruption events. (i) Following the disruption of the replication machinery (grayed circles) by DNA damage (∧), (ii) PriA or Rep functions in a reaction to transiently load DnaB and DnaG to prime the leading strand and then (iii) stably load DnaB and DnaG on the lagging strand (22, 27). (iv) The leading-strand primer allows for the de novo formation of an active replisome downstream from the site of disruption. (B) A model in which PriA and Rep are required by the replisome to maintain efficient replication. (i) Following disruption by DNA damage, the recovery of DNA synthesis requires that the lesion is either repaired (ii) or bypassed (iii) by translesion synthesis (not shown), as found in previous studies (13). (iv) Since PriA and Rep are needed to maintain replication in the absence of damage, PriA and Rep would also be required for an active replisome to be maintained once the replisome is reestablished and DNA synthesis resumes.Structural Insight into the DNA-Binding Mode of the Primosomal Proteins PriA, PriB, and DnaT 1
Replication restart primosome is a complex dynamic system that is essential for bacterial survival. This system uses various proteins to reinitiate chromosomal DNA replication to maintain genetic integrity after DNA damage. The replication restart primosome in Escherichia coli is composed of PriA helicase, PriB, PriC, DnaT, DnaC, DnaB helicase, and DnaG primase.
The assembly of the protein complexes within the forked DNA responsible for reloading the replicative DnaB helicase anywhere on the chromosome for genome duplication requires the coordination of transient biomolecular interactions. Over the last decade, investigations on the structure and mechanism of these nucleoproteins have provided considerable insight into primosome assembly. In this review, we summarize and discuss our current knowledge and recent advances on the DNA-binding mode of the primosomal proteins PriA, PriB, and DnaT.
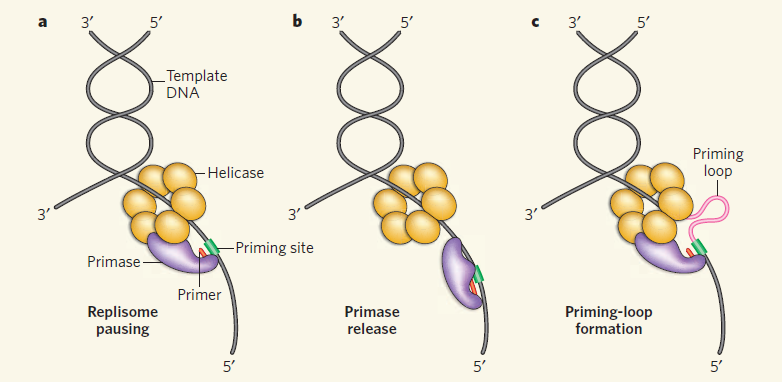
Genome integrity should be maintained from generation to generation to ensure proper cell function and survival [1–3]. In bacteria, some exogenous and endogenous sources of DNA damage can inactivate a large proportion of replication forks [4, 5]. When DNA is damaged, the replication machinery, originally initiated at oriC, can be arrested and disassembled anywhere along the DNA, leading to replication failure [5, 6]. To reload DnaB helicase for oriC-independent DNA replication, collapsed DNA replication forks must be reactivated by the replication restart primosome [7, 8]. Primosome is the protein complex responsible for the conversion of single-stranded circular DNA to the replicative-form DNA in the replication cycle of ϕX174 phage [9, 10]. After DNA repair, the replication restart primosome [11–13], a formidable enzymatic machine, can translocate along the single-stranded DNA-binding protein (SSB), unwind the duplex DNA, and prime the Okazaki fragments required for the progression of replication forks [14].
In Escherichia coli, the replication restart primosome is composed of
PriA helicase,
PriB,
PriC,
DnaB helicase,
DnaC,
DnaT, and
DnaG primase [3].
To date, two DnaB helicase-recruiting pathways are known: PriA-PriB-DnaT-DnaC-dependent and PriC-DnaC-dependent systems; the former system uses fork structures without gaps in the leading strand, whereas the latter system preferentially uses fork structures with large gaps (>5 nucleotides) in the leading strand [3]. As shown in Figure 1, PriA can bind directly and assemble a primosome on the template without gaps in the leading strand, and PriC initiates the assembly of a primosome on a fork containing gaps in the leading strand.
Figure 1: Two DnaB helicase-recruiting pathways for DNA replication restart at the stalled replication fork in vitro. The PriA-directed pathway (i.e., PriA-PriB-DnaT-DnaC-dependent reaction) preferentially uses fork structures without gaps in the leading strand, whereas the PriC-directed pathway (i.e., PriC-DnaC-dependent system) preferentially uses fork structures containing large gaps (>5 nucleotides) in the leading strand.
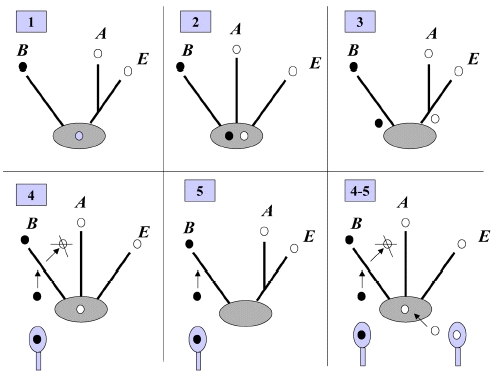
A hand-off mechanism for PriA-directed primosome assembly [15] has been proposed (Figure 2), whereby (i) PriA recognizes and binds to a replication fork; (ii) PriB joins PriA to form a PriA-PriB-DNA ternary complex; (iii) DnaT participates in this nucleocomplex to form a triprotein complex, in which PriB is released from ssDNA due to recruitment of DnaT; (iv) the PriA-PriB-DnaT-DNA quaternary complex loads the DnaB/C complex; (v) DnaB is loaded on the lagging strand template. Genetic analyses suggest that these primosomal proteins are essential replication proteins for bacterial cell growth [12, 16–21].These proteins are required for reinitiating chromosomal DNA replication in bacteria; thus, blocking their activities would be detrimental to bacterial survival [22, 23]. Several primosomal proteins, such as PriA, PriB, PriC, and DnaT, are not found in humans; Figure 2: A hand-off mechanism for the replication restart primosome assembly. The proposed assembly mechanism is as follows. (i) PriA recognizes and binds to a replication fork, (ii) PriB joins PriA to form a PriA-PriB-DNA ternary complex, (iii) DnaT participates in this nucleocomplex to form a triprotein complex, in which PriB is released from ssDNA due to recruitment of DnaT, (iv) the PriA-PriB-DnaT-DNA quaternary complex loads the DnaB/C complex, and (v) DnaB is loaded on the lagging strand template.Over the past 10 years, considerable progress has been made in the structural mechanisms of the replication restart primosome assembly. The structural information is a prerequisite for formulating any model of the assembly mechanism of the primosome (Table 1). In the following sections, we summarize and discuss our current knowledge and recent advances on the DNA-binding mode of the primosomal proteins PriA, PriB, and DnaT.
Figure 2: A hand-off mechanism for the replication restart primosome assembly. The proposed assembly mechanism is as follows. (i) PriA recognizes and binds to a replication fork, (ii) PriB joins PriA to form a PriA-PriB-DNA ternary complex, (iii) DnaT participates in this nucleocomplex to form a triprotein complex, in which PriB is released from ssDNA due to recruitment of DnaT, (iv) the PriA-PriB-DnaT-DNA quaternary complex loads the DnaB/C complex, and (v) DnaB is loaded on the lagging strand template.Over the past 10 years, considerable progress has been made in the structural mechanisms of the replication restart primosome assembly. The structural information is a prerequisite for formulating any model of the assembly mechanism of the primosome (Table 1). In the following sections, we summarize and discuss our current knowledge and recent advances on the DNA-binding mode of the primosomal proteins PriA, PriB, and DnaT.
PriA HelicasePriA functions as a scaffold that recruits other primosomal proteins. It was originally discovered as an essential factor for the conversion of single-stranded circular DNA to the replicative-form DNA of ϕX174 single-stranded phage in vitro [27]. The priA mutant of E. coli exhibits complex phenotypes that include reduced viability, chronic induction of SOS response, rich media sensitivity, decreased homologous recombination, sensitivity to UV irradiation, defective double-stranded break repair, and both induced and constitutive stable DNA replication [6, 12, 28–30]. The native PriA is a monomer with a molecular mass of ~82 kDa. The tertiary structure of the monomer contains two functional domains, namely, the helicase domain (HD), which encompasses ~540 amino acid residues from the C-terminus, and the DNA-binding domain, which comprises ~181 amino acid residues from the N-terminus [31–33]. PriA is a DEXH-type helicase that unwinds DNA with a 3′ to 5′ polarity [34]. Fuelled by the binding and hydrolysis of ATP, PriA moves along the nucleic acid filaments with other primosomal proteins and separates double-stranded DNA into their complementary single strands [35]. PriA preferentially binds to a D-loop-like structure by recognizing a bend at the three-way branched DNA structures and duplex DNA with a protruding 3′ single strand [32, 36, 37]. PriA interacts with SSB [38], PriB [15, 39, 40], and DnaT [15]. PriA can unwind the nascent lagging strand DNA to create a suitable binding site to help PriC load the DnaB helicase onto stalled replication forks where a gap exists in the nascent leading strand [41, 42]. The crystal structures of the N-terminal 105 amino acid residue segment of E. coli PriA (EcPriA) in complex with different deoxydinucleotides show a feasible interaction model for the base-non-selective recognition of the 3′-terminus of DNA between the nucleobase and the DNA-binding sites of EcPriA [43].
PriA helicase and SSB interact physically and functionally 2
PriA helicase is the major DNA replication restart initiator in Escherichia coli and acts to reload the replicative helicase DnaB back onto the chromosome at repaired replication forks and D-loops formed by recombination. We have discovered that PriA-catalysed unwinding of branched DNA substrates is stimulated specifically by contact with the single-strand DNA binding protein of E.coli, SSB. This stimulation requires binding of SSB to the initial DNA substrate and is effected via a physical interaction between PriA and the C-terminus of SSB. Stimulation of PriA by the SSB C-terminus may act to ensure that efficient PriA-catalysed reloading of DnaB occurs only onto the lagging strand template of repaired forks and D-loops.INTRODUCTION
Genome duplication presents a formidable enzymatic challenge, requiring the high fidelity replication of millions of bases of DNA. Moreover, DNA replication occurs in a complex environment. The template is an inherently unstable polymer subject to a constant barrage of chemical insults (1), whilst conflicts between replication and other essential processes such as transcription are unavoidable (2–4). As a result, replication forks may stall frequently and require some form of repair to allow completion of chromosomal duplication (5,6). Failure to solve these replicative problems comes at a high price, with the consequences being genome instability, cell death and, in higher organisms, cancer. Prokaryotic studies have highlighted the central role played by recombination enzymes in fork repair (7). Damaged replication forks appear to have two fates in Escherichia coli. First, they may be processed so that the original blocking lesion is removed or bypassed, and replication resumed once the replicative machinery has been reloaded back onto the DNA fork structure (3,8,9). Second, stalled replication forks may break to leave one intact duplex and a free duplex DNA end (9–11). Recombination of the free duplex end with the intact sister duplex creates a D-loop onto which the replication machinery can be reloaded (12).In both proposed replication repair pathways, the final stage of repair requires the restart of DNA replication. The key to initiation of DNA replication is loading of the replicative helicase DnaB onto ssDNA. DnaB catalyses unwinding of the parental DNA strands (13) and facilitates assembly of the remaining components of the replisome (14). Loading of DnaB during initiation of chromosomal duplication in E.coli is catalysed by DnaA in a tightly regulated manner at the start of the cell cycle and at a specific locus within the chromosome, oriC (15). In contrast, replication fork repair and hence reloading of DnaB may be needed away from oriC at any point within the chromosome and at any stage during chromosomal duplication. The potentially catastrophic effects of uncontrolled initiation of chromosomal duplication on genome stability suggests that replication restart must be regulated as tightly as DnaA-directed replication initiation at oriC. This implies reloading of DnaB must occur only on ssDNA at repaired forks or D-loops rather than onto other regions of ssDNA, such as those created by blocks to lagging strand synthesis (16,17). Thus an alternative replication initiator protein, PriA helicase, is utilized during replication restart to reload DnaB back onto the chromosome (18).The requirement to reload DnaB only onto repaired forks and D-loops is thought to be reflected in the preferential binding of PriA to branched DNA structures in vitro (19,20). At such structures, PriA displays two activities. PriA facilitates loading of DnaB onto the lagging strand template via a complex series of protein–protein interactions involving PriB, PriC and DnaT (21–24). However, DnaB can bind only to ssDNA (13). Thus the second enzymatic function of PriA, a 3′ to 5′ DNA helicase activity (25), acts to unwind any lagging strand DNA present at the fork to generate a ssDNA binding site for DnaB (26). The importance of PriA-directed replication restart is underlined by the decrease in viability, defective homologous recombination and extreme sensitivity to exposure to DNA damaging agents exhibited by priA null strains (27–29). There exists also an alternative pathway of replication restart that is not dependent on PriA but on Rep helicase (24). Although rep mutants do not show the extreme phenotypes displayed by priA defective cells (30), rep and priA mutations are synthetically lethal (9,24). This suggests that Rep helicase may provide an accessory replication repair function. However, molecular details of the interplay between PriA- and Rep-dependent replication repair pathways remain unknown.Here we show that SSB stimulates PriA-catalysed unwinding of branched DNA substrates. This stimulation requires binding of SSB to the initial DNA substrate and contact between PriA and the C-terminus of SSB. In contrast, neither a physical nor a functional interaction was detected between SSB and Rep helicase. A mutation within the C-terminus of SSB impairs interaction with PriA in vitro, and correlates with the DNA repair and recombination defects seen in strains carrying this ssb mutation. Contact between SSB and PriA may therefore play a critical role in coordinating reloading of the replisome at repaired forks and D-loops.Crystal structure of DnaT 84–153-dT10 ssDNA complex reveals a novel single-stranded DNA binding mode 3
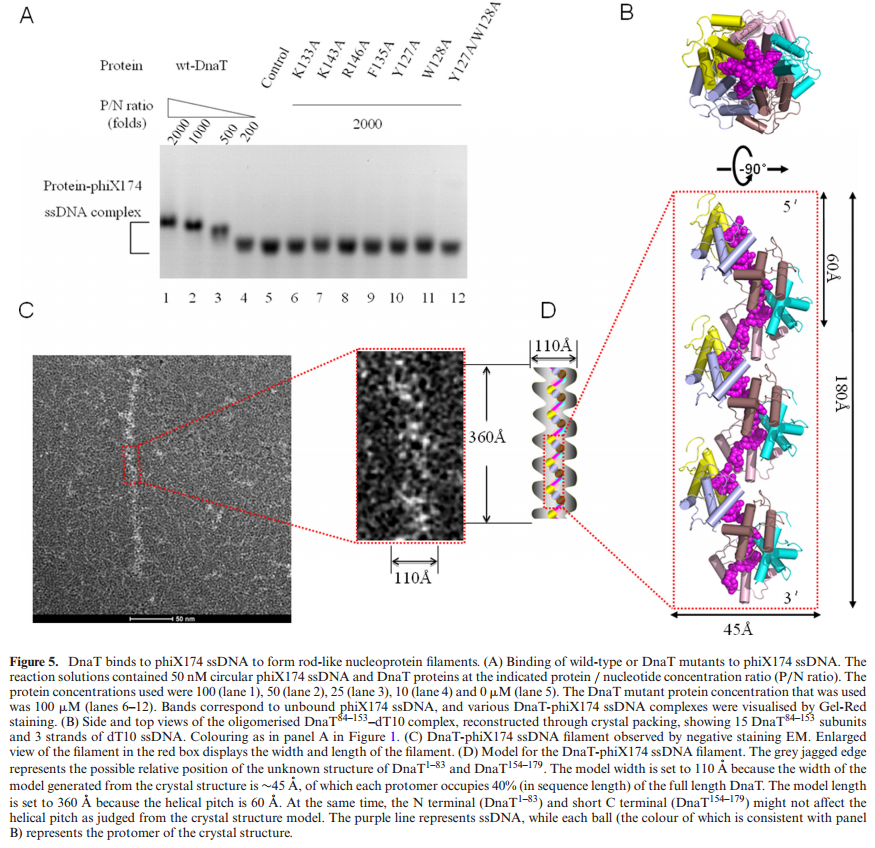
ABSTRACTDnaT is a primosomal protein that is required for the stalled replication fork restart in Escherichia coli. As an adapter, DnaT mediates the PriA-PriB-ssDNA ternary complex and the DnaB/C complex. However, the fundamental function of DnaT during PriAdependent primosome assembly is still a black box. Here, we report the 2.83 A DnaT ˚ 84–153-dT10 ssDNA complex structure, which reveals a novel three-helix bundle single-stranded DNA binding mode. Based on binding assays and negative-staining electron microscopy
results, we found that DnaT can bind to phiX 174 ssDNA to form nucleoprotein filaments for the first time, which indicates that DnaT might function as a scaffold protein during the PriA-dependent primosome assembly. In combination with biochemical analysis, we propose a cooperative mechanism for the binding of DnaT to ssDNA and a possible model for the assembly of PriA-PriB-ssDNA-DnaT complex that sheds light on the function of DnaT during the primosome assembly and stalled replication fork restart. This report presents the first structure of the DnaT C-terminal complex with ssDNA and a novel model that explains the interactions between the three-helix bundle and ssDNA.


1) http://www.hindawi.com/journals/bmri/2014/195162/
2) http://www.ncbi.nlm.nih.gov/pmc/articles/PMC535688/
3) http://nar.oxfordjournals.org/content/early/2014/07/22/nar.gku633.full.pdf
4) http://jb.asm.org/content/194/15/3977.full
5) http://web.pdx.edu/~justc/papers/recJrecQpaper.pdf
6) http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4260596/
7) http://europepmc.org/articles/PMC3710785
8 ) https://en.wikipedia.org/wiki/RecQ_helicase
9) http://www.bio.davidson.edu/Courses/Molbio/MolStudents/spring2003/Baxter/BLMgene.html
10) http://www.ebi.ac.uk/interpro/downloads/protein/prot_foc_12_06.pdf
11) http://www.nature.com/nrc/journal/v3/n3/full/nrc1012.html
12) http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1373692/