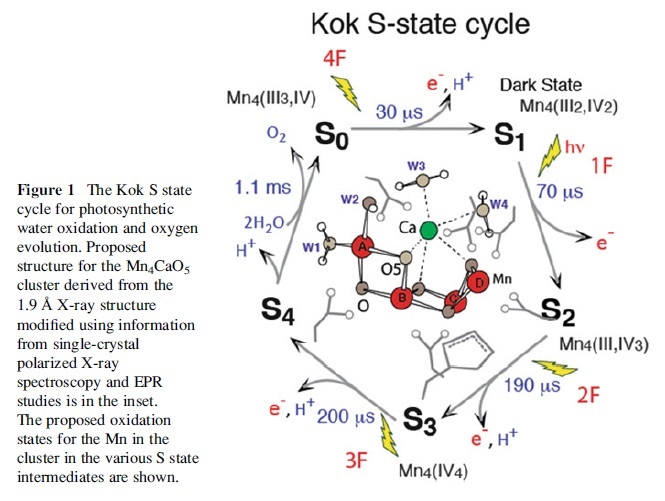
The oxygen evolving complex (OEC) of photosystem II is irreducible complex.
https://reasonandscience.catsboard.com/t1583-the-oxygen-evolving-complex-oec-of-photosystem-ii-is-irreducible-complex
1. One of the most important and fundamental biochemical reactions on which all advanced life-forms depend is performed by the oxygen-evolving complex (OEC) in oxygenic photosynthesis, responsible for catalyzing the light-driven oxidation of water to molecular oxygen in plants, algae, and cyanobacteria. It is also described as "undoubtedly one of the most remarkable inventions in all of biology." OEC is surrounded by 4 core proteins of photosystem II at the membrane-lumen interface. It remains a fundamental mystery of how this complicated, four-electron transfer process originated. Enigmatic is how the precise geometry and unique mechanism of the OEC came about. Key differences exist between oxygenic and anoxygenic photosynthetic machinery with no apparent homologs or transitional forms that would provide clues to their development. Foremost among these differences is the presence and key role of manganese at the site of water oxidation in photosystem II. This is distinct from bacterial anoxygenic reaction centers, which rely on redox-active periplasmic proteins as electron donors.
2. According to peer-reviewed scientific papers, each of the four extrinsic proteins, (PsbO, PsbP, PsbQ, and PsbR) of plants are ESSENTIAL, and each was tested upon mutated form, and the mechanism was found inefficient and compromising the OEC function. Furthermore, a water network around the Mn4CaO5 cluster and D1 protein subunit of PSII is also indispensable, and irreducible. Site-directed mutants show severe impairment of the water oxidation cycle and fail to grow photoautotrophically.
3. That means evolutionary intermediates are non-functional. There is a precise fit and size matching of the residues with the individual atoms of the clusters. This is evidence, that this most fundamental biochemical reaction could not have emerged by evolutionary, step-wise mechanisms, and therefore, Darwin's theory has been falsified and refuted. The only plausible alternative to darwinian evolution is intelligent design.
In leaves, there is a special assembly called Photosystem II (named because it was discovered second). A photon strikes this, and it is channeled into a type of chlorophyll called P680. There it knocks out an electron from an atom, and this energetic electron eventually helps make sugars from CO2. But then, the P680 must replenish the lost electron. This is a big problem for artificial photosynthesis—human chemists have also so far been unable to produce a system that replenishes the electrons knocked out by the photons. Photosynthesis would have quickly ground to a halt without this, so how are the electrons replaced?
They come from a special catalytic core, which removes the required electrons from water, again with the help of light. The light breaks two molecules of water into a molecule of oxygen, four electrons and two hydrogen ions.
The core has a unique arrangement of atoms, with an unusual cube of three atoms of Mn, one Ca and four O, attached to a single Mn [update: see Where Water Is Oxidized to Dioxygen: Structure of the Photosynthetic Mn4Ca Cluster, Science 314(5800):821–825, 3 November 2006; Learning how nature splits water]. This core builds up enough energy, in the form of redox potential,8 in stages by absorbing four photons.
The redox potential of water is +2.5 V, while each photon raises the catalytic core’s redox potential by 1 V. So after the third stage, there is enough energy for the single Mn to remove an electron from a water molecule, leaving an OH radical and H+ ion. Then the catalytic core gets to the fourth stage, and provides the Mn atom with enough power to attack the OH radical and leaves a highly reactive O atom and another H+ ion. At this moment, the Ca atom in the cube plays its essential role. It is holding another water molecule in just the right place, so it can be attacked by this O atom, producing an O2 molecule, two more H+ ions and two electrons.The unique Mn3CaO4–Mn arrangement is present in all plants, algae and cyanobacteria, which suggests that this arrangement is essential. Not surprising, because it must be able to store the energy from four photons, and hold water molecules in just the right positions. This structure had to be complete otherwise it would not work at all—in splitting water and replenishing electrons. Therefore it could not be built up gradually by small changes by natural selection. This is because an incomplete intermediate system is no use at all, so it would not be selected.
And even this core would be useless without many other coordinated features. For example, as above, the energy involved is damaging for biological molecules. Yet there are key proteins required, but must be constantly repaired, so these mechanisms must be in place too. In fact, instability of these proteins made it hard to work out the core’s structure.9
If the most intelligent human designers can’t duplicate photosynthesis, then it’s perfectly scientific to believe that photosynthesis had a far more intelligent designer. This is especially so since Darwinian processes could not have generated photosynthesis, because there are too many intricate mechanisms necessary for it to work at all.
Plants were there right at the beginning
The core of the water-splitting complex is a heterodimer of two closely related proteins, namely D1 and D2. D1 is among the most highly conserved proteins in nature It works for approximately 10 000 electron transfers and then becomes irreversibly damaged. The damage leads to a loss of photosynthetic capacity, especially at high light under nutrient-limiting conditions. 16
Why has not nature developed a better oxygen-evolving machine? We suggest that the answer is: ‘Because it can't’. D1 is one of the most conserved proteins in the photosynthetic apparatus; the sequences are more than 80% identical between cyanobacteria and higher plants, and approximately 90% similar. The extraordinary conservation of this protein, which is irreversibly damaged by a product of its own catalysis, suggests that the restrictions imposed by protein–protein, protein–prosthetic group and protein–lipid interactions cannot be overcome.
There have been literally thousands of papers discussing the mechanism of the degradation of D1 in vivo and while the exact mechanism for the process remains controversial and the repair mechanism remains almost completely unknown, it is clear that the production of a reactive oxygen species, an ‘unintended’ consequence of the production and consumption of O2, is almost certainly the proximal cause.
It takes a large amount of energy to split water into hydrogen and oxygen. This is because oxygen really wants to acquire the two extra electrons necessary to fill its outer shell, and hydrogen is a relatively easy place to get them. This, in turn, means that water is a very stable molecule, and it, therefore, takes a lot of effort – in this case, the power of a car battery – to split it apart. The structure of the Oxygen Evolving Complex was
determined only in 2006, and it is only in the last few years that the locations of each of the 46,630 atoms in photosystem II have been mapped.
"Of all the biochemical inventions in the history of life, the machinery to oxidize water — photosystem II — using sunlight is surely one of the grandest." (Sessions, A. et al, 2009)
The overwhelming source ofO2 on Earth is photobiological oxidation of water; neither the evolution nor the mechanism of this process is completely understood. Apparently, it arose once in a single clade of bacteria and was then appropriated via a single event, in which one cell engulfed another (endosymbiosis) to form a new symbiotic organism. The core of the oxidation machinery is photosystem II, a large protein complex containing four manganese atoms that are photocatalytically oxidized to create electron holes upstream.
Deep in the thylakoid membrane, the key process that produces breathable oxygen in the Earth’s atmosphere takes place, namely, the splitting of water molecules and the subsequent release of molecular oxygen. Without the OEC, no advanced life on earth would be possible. This process, which occurs in the oxygen-evolving complex (OEC) of the intricately arranged multiprotein and pigment complex known as photosystem II (PSII), is driven by the strong electrochemical potential generated from the capture and conversion of visible light to chemical energy. First, chlorophyll molecules in PSII absorb photons and lose electrons. These electrons are then passed through an electron transport chain, creating redox potential strong enough to oxidize water, which leads to the evolution of molecular oxygen and the release of protons into the thylakoid lumen. This proton gradient is a major contributor to the proton motive force (PMF), which is used to biosynthesize ATP from ADP via ATP synthase.
The OEC contains an inorganic, flexible Mn4CaO5 cluster resembling a distorted chair, which is bound to a pocket formed by six amino acids from the D1 core subunit protein and one amino acid from the light-harvesting protein CP43 . The Mn4CaO5 cluster is stabilized, at least in part, by the PsBO subunits of the OEC. The OEC is composed of a cluster of manganese, calcium and chloride ions bound to extrinsic proteins. In cyanobacteria there are five extrinsic proteins in OEC (PsbO, PsbP-like, PsbQ-like, PsbU and PsbV), while in plants there are only three (PsbO, PsbP and PsbQ). Maintenance of the highly dynamic Mn4CaO5 cluster also requires the delivery of a constant supply of the proper levels of Mn2+ and Ca2+.
The functional features of PSII in all oxygenic photosynthetic organisms are remarkably similar. The mechanism of water oxidation has remained virtually unchanged between green plants and cyanobacteria, and is similar in all higher plants. Studies on PSII from plants, algae, and cyanobacteria have revealed several PSII proteins that collectively regulate the unique redox environment of this inorganic catalytic center 12
Mainstream scientific papers quoted and cited below state that each of the extrinsic proteins, (PsbO, PsbP, PsbQ and PsbR) of plants are ESSENTIAL, and each was tested upon mutated form, and the mechanism was found inefficient, and compromising the OEC function. Furthermore, a water network around the Mn4CaO5 cluster, and D1 protein subunit of PSII are also indispensable, and irreducible.
PsbO
PsbO appears to be the most important extrinsic protein for oxygen evolution. PsbO lies closest to the Mn cluster where water oxidation occurs, and has a stabilising effect on the Mn cluster. As a result, PsbO is often referred to as the Mn-stabilising protein (MSP), although none of its amino acids are likely ligands for Mn. Calcium ions were found to modify the conformation of PsbO in solution
The photosystem II (PSII) manganese-stabilizing protein (PsbO) is known to be the essential PSII extrinsic subunit for stabilization and retention of the Mn and Cl(-) cofactors in the oxygen evolving complex (OEC) of PSII, but its function relative to Ca(2+) is less clear.
What happens if PsbO is mutated ?
The data presented here show that Asn, Glu, or Lys mutations in PsbO-Asp157 modify PsbO thermostability in solution, which is consistent with the previously reported perturbation of the functional assembly of PsbO-Asp157 mutants into PSII that caused inefficient Cl(-) retention by PSII.
PsbP
This family represents the PSII OEC protein PsbP. Both PsbP and PsbQ are regulators that are necessary for the biogenesis of optically active PSII. PsbP increases the affinity of the water oxidation site for chloride ions and provides the conditions required for high affinity binding of calcium ions [PMID: 9039496,PMID: 8910540]. The crystal structure of PsbP from Nicotiana tabacum (Common tobacco) revealed a two-domain structure, where domain 1 may play a role in the ion retention activity in PSII, the N-terminal residues being essential for calcium and chloride ion retention activity [PMID: 15031714]. PsbP is encoded in the nuclear genome in plants.
What happens if PsbO is mutated ?
these mutants show striking plant developmental and morphological defects. 4
These data indicate that assembly and/or maintenance of the functional MnCa cluster is perturbed in absence of PsbP, which impairs accumulation of final active forms of PSII supercomplexes. 5
Both psbPand psbQ inactivation mutants exhibited reduced photoautotrophic growth as well as decreased water oxidation activity under CaCl2-depleted conditions. 11
PsbQ
This family represents the PSII OEC protein PsbQ. Both PsbQ and PsbP (IPR002683) are regulators that are necessary for the biogenesis of optically active PSII. The crystal structure of PsbQ from spinach revealed a 4-helical bundle polypeptide. The distribution of positive and negative charges on the protein surface might explain the ability of PsbQ to increase the binding of chloride and calcium ions and make them available to PSII 6.
PsbP and PsbQ proteins are extrinsic subunits of photosystem II (PSII) and optimize the oxygen evolution reaction by regulating the binding properties of the essential cofactors Ca2 + and Cl−. These data suggest that the major function of PsbQ is to stabilize PsbP binding, thereby contributing to the maintenance of the catalytic Mn cluster of the water oxidation machinery in higher plant PSII. 8
What happens if PsbO is mutated ?
plants lacking PsbR or both PsbR and PsbQ are characterized by more pronounced defects in PSII activity. 7
PsbR 1
Additionally, the PsbR protein has been found in plant PSII and anticipated to play a role in water oxidation, yet the physiological significance of PsbR has remained obscure.
What happens if PsbO is mutated ?
Using the Arabidopsis psbR mutant, we showed that the light-saturated rate of oxygen evolution is strongly reduced in the absence of PsbR, particularly in low light-grown plants.
Further essential parts :
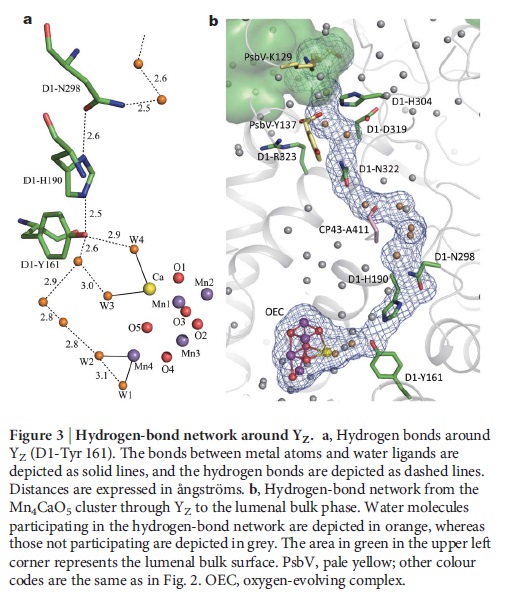
Role of a Water Network around the Mn4CaO5 Cluster in Photosynthetic Water Oxidation: 9
Around the Mn4CaO5 cluster, a hydrogen bond network is formed by several water molecules, including four water ligands.
These results suggest that the water network around the Mn4CaO5 cluster plays an essential role in the water oxidation mechanism particularly in a concerted process of proton transfer and water insertion during the S2 → S3 transition.
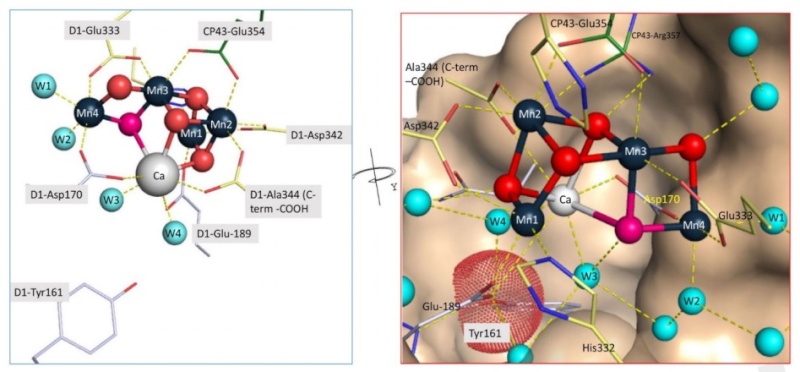
The reaction center cofactors involved in charge separation and water oxidation are coordinated by a pair of homologous protein subunits, known as D1 (PsbA) and D2 (PsbD). The D1 protein provides most of the ligands to the Mn4CaO5 cluster where water oxidation occurs. 10
Residue E354 of the CP43 coordinates Mn3 and Mn2 of the Mn4CaO5 cluster and R357 offers a hydrogen bond to O2 and O4 11
Direct ligation of the manganese ions appears to come from at least five amino acid residues of the D1 protein and one residue of the CP43 subunit 12
What happens if the two residues are mutated?
Site-directed mutants of these two residues show a severe impairment of the water oxidation cycle and fail to grow photoautotrophically.
That means evolutionary intermediates are non-functional. As we can see, there is a precise fit and size matching of the residues with the individual atoms of the clusters. How was this precision achieved? trial and error ?
In addition to this, both CP43 and CP47 play important roles in water and proton access into or out of the cluster
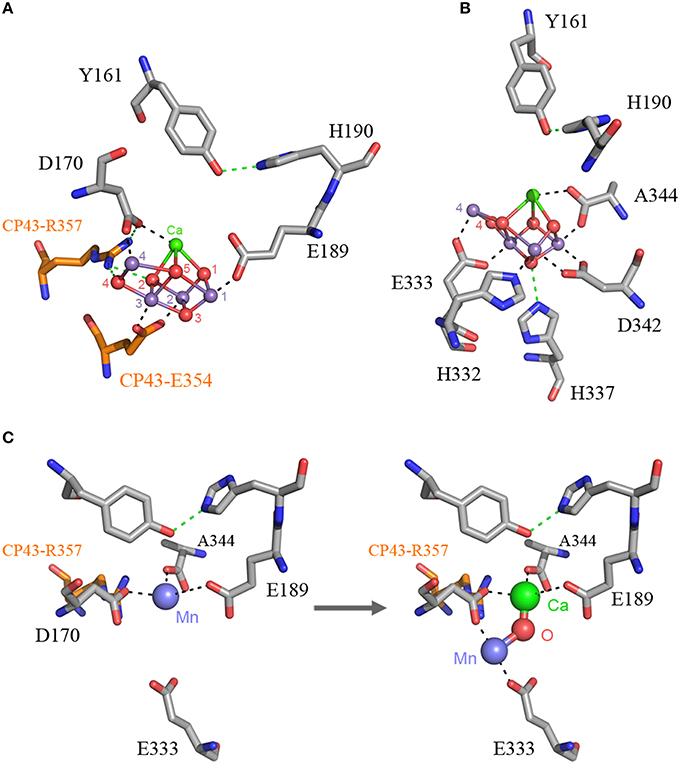
Figure 2. The Mn4CaO5 cluster of Photosystem II as resolved in the crystal structure
Panel (A) shows the cluster coordinated by the inner ligands D170 and E189 and the ligands provided by the CP43 subunit, E354, and R357.
Panel (B) shows the ligands provided from the C-terminus of the D1 protein.
Panel (C) shows a proposal for the high-affinity Mn binding site. After oxidation of the first Mn(II) to Mn(III), which might occur concomitantly with the deprotonation of a ligating water molecule, Ca2+ binds. The binding of Ca2+ shifts the initially bound Mn(III) to a position similar to that of Mn4 in the intact cluster.
Anthony W. Larkum Early Archean origin of Photosystem II 09 November 2018
How and when Type II reaction centers diversified, and how and when one of these reaction centers evolved the capacity to oxidize water are questions that still remain to be answered.
https://onlinelibrary.wiley.com/doi/10.1111/gbi.12322
1) http://www.jbc.org/content/281/1/145.full
2) http://www.life.illinois.edu/govindjee/Electronic%20Publications/2011/2011_Najafpour_Gocindjee.pdf
3) http://www.ebi.ac.uk/interpro/entry/IPR002628
4) http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028624
5) http://www.sciencedirect.com/science/article/pii/S0005272809000929
6) http://www.ebi.ac.uk/interpro/entry/IPR008797
7) http://www.ncbi.nlm.nih.gov/pubmed/23647309
8 http://www.sciencedirect.com/science/article/pii/S0005272812000114
9) http://www.ncbi.nlm.nih.gov/pubmed/26716470
10) http://mbe.oxfordjournals.org/content/32/5/1310.full
11) http://journal.frontiersin.org/article/10.3389/fpls.2016.00257/full
12) http://www.ncbi.nlm.nih.gov/pmc/articles/PMC519205/
13) http://www.plantcell.org/content/early/2016/04/06/tpc.16.00269.full.pdf
14) http://science.sciencemag.org.sci-hub.cc/content/322/5901/540.full
15 ) Brian Cox, Wonders of life, page 37
16) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2606772/#fn2
17. https://creation.com/green-power-photosynthesis
18. https://sci-hub.tw/https://www.sciencedirect.com/science/article/abs/pii/S0010854507001877#:~:text=The%20invention%20of%20oxygenic%20photosynthesis,history%20of%20life%20on%20Earth.&text=This%20is%20distinct%20from%20bacterial,periplasmic%20proteins%20as%20electron%20donors.
https://reasonandscience.catsboard.com/t1583-the-oxygen-evolving-complex-oec-of-photosystem-ii-is-irreducible-complex
1. One of the most important and fundamental biochemical reactions on which all advanced life-forms depend is performed by the oxygen-evolving complex (OEC) in oxygenic photosynthesis, responsible for catalyzing the light-driven oxidation of water to molecular oxygen in plants, algae, and cyanobacteria. It is also described as "undoubtedly one of the most remarkable inventions in all of biology." OEC is surrounded by 4 core proteins of photosystem II at the membrane-lumen interface. It remains a fundamental mystery of how this complicated, four-electron transfer process originated. Enigmatic is how the precise geometry and unique mechanism of the OEC came about. Key differences exist between oxygenic and anoxygenic photosynthetic machinery with no apparent homologs or transitional forms that would provide clues to their development. Foremost among these differences is the presence and key role of manganese at the site of water oxidation in photosystem II. This is distinct from bacterial anoxygenic reaction centers, which rely on redox-active periplasmic proteins as electron donors.
2. According to peer-reviewed scientific papers, each of the four extrinsic proteins, (PsbO, PsbP, PsbQ, and PsbR) of plants are ESSENTIAL, and each was tested upon mutated form, and the mechanism was found inefficient and compromising the OEC function. Furthermore, a water network around the Mn4CaO5 cluster and D1 protein subunit of PSII is also indispensable, and irreducible. Site-directed mutants show severe impairment of the water oxidation cycle and fail to grow photoautotrophically.
3. That means evolutionary intermediates are non-functional. There is a precise fit and size matching of the residues with the individual atoms of the clusters. This is evidence, that this most fundamental biochemical reaction could not have emerged by evolutionary, step-wise mechanisms, and therefore, Darwin's theory has been falsified and refuted. The only plausible alternative to darwinian evolution is intelligent design.
In leaves, there is a special assembly called Photosystem II (named because it was discovered second). A photon strikes this, and it is channeled into a type of chlorophyll called P680. There it knocks out an electron from an atom, and this energetic electron eventually helps make sugars from CO2. But then, the P680 must replenish the lost electron. This is a big problem for artificial photosynthesis—human chemists have also so far been unable to produce a system that replenishes the electrons knocked out by the photons. Photosynthesis would have quickly ground to a halt without this, so how are the electrons replaced?
They come from a special catalytic core, which removes the required electrons from water, again with the help of light. The light breaks two molecules of water into a molecule of oxygen, four electrons and two hydrogen ions.
The core has a unique arrangement of atoms, with an unusual cube of three atoms of Mn, one Ca and four O, attached to a single Mn [update: see Where Water Is Oxidized to Dioxygen: Structure of the Photosynthetic Mn4Ca Cluster, Science 314(5800):821–825, 3 November 2006; Learning how nature splits water]. This core builds up enough energy, in the form of redox potential,8 in stages by absorbing four photons.
The redox potential of water is +2.5 V, while each photon raises the catalytic core’s redox potential by 1 V. So after the third stage, there is enough energy for the single Mn to remove an electron from a water molecule, leaving an OH radical and H+ ion. Then the catalytic core gets to the fourth stage, and provides the Mn atom with enough power to attack the OH radical and leaves a highly reactive O atom and another H+ ion. At this moment, the Ca atom in the cube plays its essential role. It is holding another water molecule in just the right place, so it can be attacked by this O atom, producing an O2 molecule, two more H+ ions and two electrons.The unique Mn3CaO4–Mn arrangement is present in all plants, algae and cyanobacteria, which suggests that this arrangement is essential. Not surprising, because it must be able to store the energy from four photons, and hold water molecules in just the right positions. This structure had to be complete otherwise it would not work at all—in splitting water and replenishing electrons. Therefore it could not be built up gradually by small changes by natural selection. This is because an incomplete intermediate system is no use at all, so it would not be selected.
And even this core would be useless without many other coordinated features. For example, as above, the energy involved is damaging for biological molecules. Yet there are key proteins required, but must be constantly repaired, so these mechanisms must be in place too. In fact, instability of these proteins made it hard to work out the core’s structure.9
If the most intelligent human designers can’t duplicate photosynthesis, then it’s perfectly scientific to believe that photosynthesis had a far more intelligent designer. This is especially so since Darwinian processes could not have generated photosynthesis, because there are too many intricate mechanisms necessary for it to work at all.
Plants were there right at the beginning
The core of the water-splitting complex is a heterodimer of two closely related proteins, namely D1 and D2. D1 is among the most highly conserved proteins in nature It works for approximately 10 000 electron transfers and then becomes irreversibly damaged. The damage leads to a loss of photosynthetic capacity, especially at high light under nutrient-limiting conditions. 16
Why has not nature developed a better oxygen-evolving machine? We suggest that the answer is: ‘Because it can't’. D1 is one of the most conserved proteins in the photosynthetic apparatus; the sequences are more than 80% identical between cyanobacteria and higher plants, and approximately 90% similar. The extraordinary conservation of this protein, which is irreversibly damaged by a product of its own catalysis, suggests that the restrictions imposed by protein–protein, protein–prosthetic group and protein–lipid interactions cannot be overcome.
There have been literally thousands of papers discussing the mechanism of the degradation of D1 in vivo and while the exact mechanism for the process remains controversial and the repair mechanism remains almost completely unknown, it is clear that the production of a reactive oxygen species, an ‘unintended’ consequence of the production and consumption of O2, is almost certainly the proximal cause.
It takes a large amount of energy to split water into hydrogen and oxygen. This is because oxygen really wants to acquire the two extra electrons necessary to fill its outer shell, and hydrogen is a relatively easy place to get them. This, in turn, means that water is a very stable molecule, and it, therefore, takes a lot of effort – in this case, the power of a car battery – to split it apart. The structure of the Oxygen Evolving Complex was
determined only in 2006, and it is only in the last few years that the locations of each of the 46,630 atoms in photosystem II have been mapped.
"Of all the biochemical inventions in the history of life, the machinery to oxidize water — photosystem II — using sunlight is surely one of the grandest." (Sessions, A. et al, 2009)
The overwhelming source ofO2 on Earth is photobiological oxidation of water; neither the evolution nor the mechanism of this process is completely understood. Apparently, it arose once in a single clade of bacteria and was then appropriated via a single event, in which one cell engulfed another (endosymbiosis) to form a new symbiotic organism. The core of the oxidation machinery is photosystem II, a large protein complex containing four manganese atoms that are photocatalytically oxidized to create electron holes upstream.
Deep in the thylakoid membrane, the key process that produces breathable oxygen in the Earth’s atmosphere takes place, namely, the splitting of water molecules and the subsequent release of molecular oxygen. Without the OEC, no advanced life on earth would be possible. This process, which occurs in the oxygen-evolving complex (OEC) of the intricately arranged multiprotein and pigment complex known as photosystem II (PSII), is driven by the strong electrochemical potential generated from the capture and conversion of visible light to chemical energy. First, chlorophyll molecules in PSII absorb photons and lose electrons. These electrons are then passed through an electron transport chain, creating redox potential strong enough to oxidize water, which leads to the evolution of molecular oxygen and the release of protons into the thylakoid lumen. This proton gradient is a major contributor to the proton motive force (PMF), which is used to biosynthesize ATP from ADP via ATP synthase.
The OEC contains an inorganic, flexible Mn4CaO5 cluster resembling a distorted chair, which is bound to a pocket formed by six amino acids from the D1 core subunit protein and one amino acid from the light-harvesting protein CP43 . The Mn4CaO5 cluster is stabilized, at least in part, by the PsBO subunits of the OEC. The OEC is composed of a cluster of manganese, calcium and chloride ions bound to extrinsic proteins. In cyanobacteria there are five extrinsic proteins in OEC (PsbO, PsbP-like, PsbQ-like, PsbU and PsbV), while in plants there are only three (PsbO, PsbP and PsbQ). Maintenance of the highly dynamic Mn4CaO5 cluster also requires the delivery of a constant supply of the proper levels of Mn2+ and Ca2+.
The functional features of PSII in all oxygenic photosynthetic organisms are remarkably similar. The mechanism of water oxidation has remained virtually unchanged between green plants and cyanobacteria, and is similar in all higher plants. Studies on PSII from plants, algae, and cyanobacteria have revealed several PSII proteins that collectively regulate the unique redox environment of this inorganic catalytic center 12
Mainstream scientific papers quoted and cited below state that each of the extrinsic proteins, (PsbO, PsbP, PsbQ and PsbR) of plants are ESSENTIAL, and each was tested upon mutated form, and the mechanism was found inefficient, and compromising the OEC function. Furthermore, a water network around the Mn4CaO5 cluster, and D1 protein subunit of PSII are also indispensable, and irreducible.
PsbO
PsbO appears to be the most important extrinsic protein for oxygen evolution. PsbO lies closest to the Mn cluster where water oxidation occurs, and has a stabilising effect on the Mn cluster. As a result, PsbO is often referred to as the Mn-stabilising protein (MSP), although none of its amino acids are likely ligands for Mn. Calcium ions were found to modify the conformation of PsbO in solution
The photosystem II (PSII) manganese-stabilizing protein (PsbO) is known to be the essential PSII extrinsic subunit for stabilization and retention of the Mn and Cl(-) cofactors in the oxygen evolving complex (OEC) of PSII, but its function relative to Ca(2+) is less clear.
What happens if PsbO is mutated ?
The data presented here show that Asn, Glu, or Lys mutations in PsbO-Asp157 modify PsbO thermostability in solution, which is consistent with the previously reported perturbation of the functional assembly of PsbO-Asp157 mutants into PSII that caused inefficient Cl(-) retention by PSII.
PsbP
This family represents the PSII OEC protein PsbP. Both PsbP and PsbQ are regulators that are necessary for the biogenesis of optically active PSII. PsbP increases the affinity of the water oxidation site for chloride ions and provides the conditions required for high affinity binding of calcium ions [PMID: 9039496,PMID: 8910540]. The crystal structure of PsbP from Nicotiana tabacum (Common tobacco) revealed a two-domain structure, where domain 1 may play a role in the ion retention activity in PSII, the N-terminal residues being essential for calcium and chloride ion retention activity [PMID: 15031714]. PsbP is encoded in the nuclear genome in plants.
What happens if PsbO is mutated ?
these mutants show striking plant developmental and morphological defects. 4
These data indicate that assembly and/or maintenance of the functional MnCa cluster is perturbed in absence of PsbP, which impairs accumulation of final active forms of PSII supercomplexes. 5
Both psbPand psbQ inactivation mutants exhibited reduced photoautotrophic growth as well as decreased water oxidation activity under CaCl2-depleted conditions. 11
PsbQ
This family represents the PSII OEC protein PsbQ. Both PsbQ and PsbP (IPR002683) are regulators that are necessary for the biogenesis of optically active PSII. The crystal structure of PsbQ from spinach revealed a 4-helical bundle polypeptide. The distribution of positive and negative charges on the protein surface might explain the ability of PsbQ to increase the binding of chloride and calcium ions and make them available to PSII 6.
PsbP and PsbQ proteins are extrinsic subunits of photosystem II (PSII) and optimize the oxygen evolution reaction by regulating the binding properties of the essential cofactors Ca2 + and Cl−. These data suggest that the major function of PsbQ is to stabilize PsbP binding, thereby contributing to the maintenance of the catalytic Mn cluster of the water oxidation machinery in higher plant PSII. 8
What happens if PsbO is mutated ?
plants lacking PsbR or both PsbR and PsbQ are characterized by more pronounced defects in PSII activity. 7
PsbR 1
Additionally, the PsbR protein has been found in plant PSII and anticipated to play a role in water oxidation, yet the physiological significance of PsbR has remained obscure.
What happens if PsbO is mutated ?
Using the Arabidopsis psbR mutant, we showed that the light-saturated rate of oxygen evolution is strongly reduced in the absence of PsbR, particularly in low light-grown plants.
Further essential parts :
Role of a Water Network around the Mn4CaO5 Cluster in Photosynthetic Water Oxidation: 9
Around the Mn4CaO5 cluster, a hydrogen bond network is formed by several water molecules, including four water ligands.
These results suggest that the water network around the Mn4CaO5 cluster plays an essential role in the water oxidation mechanism particularly in a concerted process of proton transfer and water insertion during the S2 → S3 transition.
The reaction center cofactors involved in charge separation and water oxidation are coordinated by a pair of homologous protein subunits, known as D1 (PsbA) and D2 (PsbD). The D1 protein provides most of the ligands to the Mn4CaO5 cluster where water oxidation occurs. 10
Residue E354 of the CP43 coordinates Mn3 and Mn2 of the Mn4CaO5 cluster and R357 offers a hydrogen bond to O2 and O4 11
Direct ligation of the manganese ions appears to come from at least five amino acid residues of the D1 protein and one residue of the CP43 subunit 12
What happens if the two residues are mutated?
Site-directed mutants of these two residues show a severe impairment of the water oxidation cycle and fail to grow photoautotrophically.
That means evolutionary intermediates are non-functional. As we can see, there is a precise fit and size matching of the residues with the individual atoms of the clusters. How was this precision achieved? trial and error ?
In addition to this, both CP43 and CP47 play important roles in water and proton access into or out of the cluster

Figure 2. The Mn4CaO5 cluster of Photosystem II as resolved in the crystal structure
Panel (A) shows the cluster coordinated by the inner ligands D170 and E189 and the ligands provided by the CP43 subunit, E354, and R357.
Panel (B) shows the ligands provided from the C-terminus of the D1 protein.
Panel (C) shows a proposal for the high-affinity Mn binding site. After oxidation of the first Mn(II) to Mn(III), which might occur concomitantly with the deprotonation of a ligating water molecule, Ca2+ binds. The binding of Ca2+ shifts the initially bound Mn(III) to a position similar to that of Mn4 in the intact cluster.
Anthony W. Larkum Early Archean origin of Photosystem II 09 November 2018
How and when Type II reaction centers diversified, and how and when one of these reaction centers evolved the capacity to oxidize water are questions that still remain to be answered.
https://onlinelibrary.wiley.com/doi/10.1111/gbi.12322
1) http://www.jbc.org/content/281/1/145.full
2) http://www.life.illinois.edu/govindjee/Electronic%20Publications/2011/2011_Najafpour_Gocindjee.pdf
3) http://www.ebi.ac.uk/interpro/entry/IPR002628
4) http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028624
5) http://www.sciencedirect.com/science/article/pii/S0005272809000929
6) http://www.ebi.ac.uk/interpro/entry/IPR008797
7) http://www.ncbi.nlm.nih.gov/pubmed/23647309
8 http://www.sciencedirect.com/science/article/pii/S0005272812000114
9) http://www.ncbi.nlm.nih.gov/pubmed/26716470
10) http://mbe.oxfordjournals.org/content/32/5/1310.full
11) http://journal.frontiersin.org/article/10.3389/fpls.2016.00257/full
12) http://www.ncbi.nlm.nih.gov/pmc/articles/PMC519205/
13) http://www.plantcell.org/content/early/2016/04/06/tpc.16.00269.full.pdf
14) http://science.sciencemag.org.sci-hub.cc/content/322/5901/540.full
15 ) Brian Cox, Wonders of life, page 37
16) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2606772/#fn2
17. https://creation.com/green-power-photosynthesis
18. https://sci-hub.tw/https://www.sciencedirect.com/science/article/abs/pii/S0010854507001877#:~:text=The%20invention%20of%20oxygenic%20photosynthesis,history%20of%20life%20on%20Earth.&text=This%20is%20distinct%20from%20bacterial,periplasmic%20proteins%20as%20electron%20donors.
Last edited by Otangelo on Tue May 17, 2022 4:51 am; edited 29 times in total