IMP Is Converted to Adenine and Guanine Ribonucleotides
In the grand saga of nucleotide synthesis, IMP takes center stage as a precursor for two vital nucleotides—AMP and GMP. These molecular heroes, AMP and GMP, hold key roles in living systems. Let's unravel their secrets and discover the magic they bring to the stage. AMP, also known as adenosine monophosphate, is a nucleotide of great significance. Its structure is a trio of marvels—a nitrogenous base named adenine, a sugar known as ribose, and a single phosphate group. Together, they form the foundation of AMP's power. This nucleotide luminary plays an essential role in a multitude of biological processes, orchestrating cellular activities with grace and precision. Now, let's shift our gaze to GMP—guanosine monophosphate—an esteemed member of the nucleotide family. Its structure closely resembles that of AMP, with a nitrogenous base called guanine, a ribose sugar, and a single phosphate group. GMP, like its counterpart AMP, holds a pivotal position in the intricate realm of nucleic acids. It contributes to the formation of DNA and RNA, participating in the dance of life's information storage and transmission. Why were AMP and GMP chosen to be part of the extraordinary quartet of RNA and DNA? The answer lies in their structural and functional prowess. Alongside their faithful partners, T and C, they contribute to the rich diversity of base pairings, elevating the informational content of DNA and RNA to new heights. The presence of AMP and GMP in this quartet is not a coincidence but a deliberate selection. Nature, in its wisdom, has chosen these nucleotides for their ability to form stable base pairs and their seamless integration within the grand structure and function of DNA and RNA. It is through the exquisite arrangement and interplay of these nucleotides that the quartet—A, T/U, G, and C—unleashes the incredible genetic diversity and capacity for storing information witnessed in all living organisms. So, let us marvel at the beauty of AMP and GMP as they join their counterparts, T and C, in the grand tale of nucleotide synthesis. Their unique structures and roles have been carefully curated to ensure the symphony of life plays out with precision. Together, they contribute to the richness of genetic information, encapsulating the wonder and complexity of the living world.
AMP synthesis
The journey from IMP to AMP and GMP unfolds through distinct pathways, each with its own set of pivotal reactions. Let's embark on this captivating adventure and explore the remarkable transformations that take place. In the realm of AMP synthesis, the first step is orchestrated by the masterful enzyme adenylosuccinate synthase, also known as adenylosuccinase. This enzymatic maestro guides the transfer of the amino group from aspartate to IMP, forging a bond between aspartate and IMP. This reaction is fueled by the energetic dance of GTP, as it sacrifices itself through hydrolysis. GTP, a nucleotide composed of a guanine base, a ribose sugar, and three phosphate groups, is a true cellular powerhouse. It serves as an energy carrier, fueling diverse processes such as protein synthesis, signal transduction, and energy metabolism. When GTP undergoes hydrolysis, a water molecule steps onto the stage, leading to the breaking of a high-energy phosphate bond. The bond between the last two phosphate groups in GTP is cleaved, resulting in the birth of GDP and the release of inorganic phosphate (Pi). GDP, formed by the removal of a phosphate group from GTP, emerges as a transformed molecule, while Pi dances its way as a separate entity. Enzymes called GTPases are the skilled facilitators of this hydrolysis dance, playing essential roles in cellular processes like G protein signaling and protein synthesis regulation. As GTP bids adieu, it leaves behind a trail of released energy, a gift for the cell to utilize in various energy-demanding endeavors. Now, armed with adenylosuccinate, our journey ventures further into the second crucial reaction. Here, adenylosuccinate lyase takes the spotlight, gracefully removing the fumarate group from adenylosuccinate. As this transformative enzyme wields its power, a wondrous alchemy occurs—AMP is born. It's important to note that adenylosuccinate lyase also catalyzes Reaction 9 of the IMP pathway, where it removes fumarate from another intermediate. Both reactions share a common thread—they add a nitrogen atom to the molecule, with fumarate emerging as a byproduct, bidding farewell to the stage. In this enthralling tale of nucleotide synthesis, we witness the magic of transformation. AMP, once a distant dream in the realm of IMP, takes shape through the orchestrated dances of adenylosuccinate synthase and adenylosuccinate lyase. The vibrant energy of GTP fuels these reactions, guiding the steps of molecular ballet. And as fumarate bids its farewell, AMP emerges as a triumphant hero, ready to fulfill its role in the grand symphony of DNA and RNA. So, let us marvel at the wonders of these reactions as we journey from IMP to AMP and GMP. It is a dance of molecular elegance, where bonds are formed, hydrolysis unlocks energy, and transformation brings forth new life. Through these remarkable processes, the intricate world of nucleotide synthesis unveils its captivating secrets, revealing the exquisite mechanisms that underpin the dance of life.
GMP synthesis
But wait, the path to GMP from IMP takes a different course—a fascinating journey that unfolds through a separate pathway. Let's embark on this alternate adventure and witness the captivating transformations that occur along the way. Our tale begins with the dehydrogenation of IMP, a pivotal moment orchestrated by the enzyme IMP dehydrogenase. This enzymatic marvel guides the removal of hydrogen from IMP, resulting in the reduction of NAD+ and the birth of xanthosine monophosphate (XMP). XMP, the ribonucleotide of the base xanthine, emerges as a new character in our narrative. This reaction holds a subtle dance of electrons, as IMP surrenders its hydrogen atoms, leading to the transformation of NAD+. Through this exchange, XMP takes its rightful place, ready to continue the journey. Now, with XMP at the center stage, the story progresses to its climax. Here, the enzyme GMP synthetase takes the spotlight, orchestrating the transfer of the amide nitrogen from glutamine to XMP. This transformative moment marks the birth of GMP. But the magic doesn't stop there—the energy required for this transfer reaction comes from the hydrolysis of ATP. ATP, the universal energy currency of cells, undergoes a mesmerizing transformation, splitting into AMP and pyrophosphate (PPi). It is through this captivating hydrolysis that the necessary energy is harnessed to drive the transfer of the amide nitrogen from glutamine to XMP. As the ATP molecule sacrifices itself, AMP emerges, and pyrophosphate is released as a separate entity. And there it is—GMP, the result of this enchanting sequence of events. Now, armed with its new identity, GMP stands ready to fulfill its role in cellular processes. It becomes a player in the grand symphony of life, contributing to the dance of DNA and RNA, fueling the intricate mechanisms that drive the wonders of biology. In this captivating narrative of nucleotide synthesis, we witness the unique pathway from IMP to GMP. IMP dehydrogenase sets the stage with its dehydrogenation prowess, giving rise to XMP. Then, GMP synthetase takes command, conducting the transfer of the amide nitrogen from glutamine to XMP, with the energetic support of ATP hydrolysis. It is through these alchemical processes that GMP is born, ready to embark on its role in the cellular symphony. So, let us rejoice in the wonders of this journey—a path that diverges from AMP synthesis to lead us to the realm of GMP. It is a dance of molecular transformations, where bonds are forged, energy is harnessed, and new characters step onto the stage. Through these magnificent processes, the world of nucleotide synthesis reveals its secrets, unraveling the intricate mechanisms that underlie the beauty of life's dance.

Adenylosuccinate synthase (ADSS)
Adenylosuccinate synthase (ADSS) is a remarkable enzyme responsible for catalyzing the formation of adenylosuccinate—a vital intermediate in the synthesis of adenosine monophosphate (AMP). AMP, in turn, plays a pivotal role in numerous cellular processes, underscoring the significance of ADSS in the intricate machinery of life. Let us embark on a journey to explore the intricacies of this enzyme and the fascinating mechanisms by which it acquires the essential cofactors for its catalytic prowess. ADSS, with a total structure weight of 48.27 kDa (kilodaltons) and an awe-inspiring assembly of 3,553 atoms, stands as a testament to the intricacy and precision of biological machinery. Its delicate balance of atoms and subunits enables it to orchestrate the formation of adenylosuccinate—a vital stepping stone on the path to AMP. To unlock its catalytic potential, ADSS relies on the presence of divalent metal ions, particularly the versatile magnesium (Mg2+). These ions act as cofactors, deftly facilitating the enzyme's catalytic activities. But how does the simplest of cells acquire these precious metal ions? The acquisition of divalent metal ions, such as magnesium, can occur through various mechanisms. One such mechanism is passive diffusion—an elegant dance between the ions and the cell membrane. Picture a scenario where the concentration of these ions is higher outside the cell compared to within its cytoplasm. In this setting, the ions possess a powerful driving force, compelling them to traverse the cell membrane and journey into the cell's inner sanctum. Through passive diffusion, these divalent metal ions, including the esteemed magnesium, spontaneously traverse the cell membrane, propelled by the concentration gradient. They gracefully move from regions of higher concentration—found outside the cell—to areas of lower concentration nestled within its delicate confines. This captivating dance continues until a harmonious equilibrium is reached, where the concentration of these ions is equal both inside and outside the cell. What distinguishes passive diffusion is its ability to transport small, uncharged molecules or ions that can effortlessly navigate the lipid bilayer of the cell membrane. These agile particles effortlessly slip through the molecular gaps, driven solely by the forces of chance and concentration. Remarkably, this process demands no additional energy or the involvement of specialized transport proteins, allowing the ions to traverse freely. However, it is crucial to note that the elegance of passive diffusion is limited to these small, uncharged entities. Larger molecules or charged ions, with their bulk and electric charges, require more intricate means of transportation. For their triumphant passage across the cell membrane, specific channels, transporters, or pumps must come into play, guiding these larger and charged entities on their destined path. As we peel back the layers of ADSS and its quest for magnesium ions, we uncover a tale of delicate balance and captivating dance. The enzyme's role in the production of AMP, intertwined with the acquisition of essential cofactors, paints a vivid picture of the marvels hidden within the microscopic world of cells. Join me as we continue to explore the remarkable intricacies of the molecular realm, where beauty and scientific wonder coexist in perfect harmony.
In the realm of simple cells like bacteria or archaea, fascinating concentration differences take place, specifically with divalent metal ions such as magnesium (Mg2+). Let's venture into this world and explore the mechanisms that establish these differences, painting a vivid picture of cellular life. One source of concentration disparity arises from the contrasting Mg2+ ion levels between the cell's external environment and its cytoplasm. Picture a cell residing in a surrounding medium with higher concentrations of Mg2+ ions than those found within its cytoplasm. This natural variance sets the stage for a wondrous play of ion dynamics. But there's more to this tale. Simple cells possess metabolic processes that actively manipulate Mg2+ ions within their cytoplasm. Enzymes and transporters engaged in cellular metabolism rely on Mg2+ ions as cofactors, consuming them in their biochemical endeavors. Through this consumption, concentration gradients are established, further enhancing the contrast between the extracellular environment and the cell's inner sanctum. Transport systems within these cells play a key role in sculpting the concentration differences. Imagine ion channels or transporters adorning the cell membrane, acting as selective gatekeepers. They facilitate the movement of Mg2+ ions, actively transporting them into the cell or ushering them out. This dynamic interplay ensures that concentration differences persist, adding a thrilling dimension to the cellular landscape. Ah, but we must not forget the cell's membrane potential, a conductor orchestrating this ion symphony. The membrane potential, the difference in electrical charge across the cell membrane, exerts its influence on charged ions like Mg2+. This electrical potential alters the ions' movement, creating disparities between the extracellular environment and the cytoplasm. It's a delicate interplay of charges, intricately shaping the ion concentrations within the cell. In the realm of cellular homeostasis, regulatory mechanisms come into play, maintaining the delicate balance of ion concentrations, including the precious Mg2+. Ion channels, transporters, and regulatory proteins take center stage, masterfully allowing or blocking the passage of ions across the membrane. With their guidance, concentration differences persist, ensuring that the dance of ions continues harmoniously within the cell. So, let us revel in the enchantment of concentration differences in simple cells, where Mg2+ ions dictate the ebb and flow of cellular life. Through mechanisms of active accumulation, consumption, and transport, these cells establish captivating disparities. And with the aid of membrane potentials and regulatory proteins, the delicate balance of ion concentrations is preserved, ensuring the dance of life remains in perfect harmony.
Step 1: Phosphorylation of IMP
Within the realm of enzymes, there lies a captivating character known as adenylosuccinate synthase. This remarkable enzyme belongs to the esteemed class of ligases, specifically the family of transferases that are skilled in forming carbon-nitrogen bonds. Let us embark on a journey through the multistep dance orchestrated by this enchanting enzyme, as it weaves together substrates and brings forth the birth of adenylosuccinate. Our tale begins with the union of two key players: inosine monophosphate (IMP) and aspartate. Picture these molecules intertwining, their atoms aligning with precision, ready to embark on a transformation of extraordinary significance. The adenylosuccinate synthase, with its enzymatic mastery, guides the way, catalyzing a symphony of chemical reactions. Step by step, the dance unfolds. In the first movement, IMP and aspartate come together, forming an intermediate known as adenylosuccinyl-AMP (ASAMP). This intermediate bears the potential for greatness, harboring the essence of adenylosuccinate within its intricate structure. But the transformation doesn't end there. The dance continues, and with each step, the ASAMP intermediate undergoes a graceful metamorphosis. In the second movement, a remarkable rearrangement occurs, leading to the formation of an energy-rich molecule called adenosine monophosphate (AMP). This pivotal step is a crucial milestone in the de novo synthesis of AMP, a molecule of great significance in the cellular symphony of life. As we witness the unfolding of this multistep reaction, we are captivated by the precision and elegance with which adenylosuccinate synthase orchestrates the union of IMP and aspartate. Its skilled hand guides the formation of adenylosuccinate, paving the way for the subsequent formation of AMP. Each step in this enchanting dance serves a purpose, bringing us closer to the grand finale. So, let us revel in the magic of adenylosuccinate synthase, a ligase of exceptional talent. It masterfully catalyzes the formation of adenylosuccinate through a captivating multistep reaction. The fusion of IMP and aspartate unveils the hidden potential within, creating a symphony of molecules that sets the stage for the birth of AMP. It is through the expertise of this remarkable enzyme that the de novo synthesis of AMP finds its path, contributing to the harmonious dance of cellular life.
Implementing conformational change functions in the protein
In the intricate dance of molecular interactions, conformational changes hold the power to elevate binding interactions to new heights. When a ligand or substrate embraces the binding pocket of a protein, a symphony of transformations is set into motion. These changes add a touch of complexity, captivating our senses and enriching the process with a mesmerizing allure. As the ligand finds its place within the binding pocket, it whispers secrets to the protein, provoking a response. The protein, like a maestro, orchestrates its movements, shifting and rearranging to accommodate the presence of its partner. Individual amino acid residues may gracefully adjust their positions, delicately harmonizing with the ligand. In some cases, larger-scale transformations occur, where entire protein domains gracefully shift, adapting to the newfound connection. These conformational changes are born from the intimate dialogue between the ligand and specific residues within the binding pocket. Like a dance partner guiding their companion, the ligand influences the protein's structure, imbuing it with a fresh elegance. But the impact doesn't end there—these changes can ripple through the protein, transcending the immediate vicinity of the binding site. This captivating phenomenon is known as allosteric regulation, where the binding event resonates with distant regions of the protein. Through allosteric effects, the protein's activity, stability, and interactions with other molecules are transformed beyond the confines of the binding site. It's as if the protein, awakened by the ligand's touch, communicates its newfound knowledge to its surroundings. This communication between the binding site and other regions of the protein serves as a vital mechanism for regulating cellular processes, a symphony of coordination in the intricate dance of life. So, let us marvel at the wonders of conformational changes induced by ligand binding. They enrich the binding interaction, enhancing specificity and affinity. As the ligand whispers its secrets, the protein responds, reshaping itself with grace and elegance. This transformative dance captivates our imagination, as allosteric effects ripple through the protein, influencing its activity, stability, and interactions. It is through this intricate communication that the protein navigates the complex symphony of cellular processes, unveiling the grandeur of nature's choreography.
Long distant signaling through allosteric networks points to a designed setup
In the amazing world of enzymes, we encounter a remarkable phenomenon known as allosteric regulation. Within these enzymes, a symphony of communication unfolds, spanning vast distances. Imagine an intricate dance between distinct binding sites—the active site, where the substrate finds its place, and the allosteric site, a distant domain with a secret to tell. This interplay of binding events holds the key to modulating the enzyme's activity, and one fascinating example is the renowned enzyme adenylosuccinate synthase. As adenylosuccinate synthase enters the spotlight, we witness its transformation through conformational changes triggered by ligand binding at the allosteric site. This intricate ballet of motion governs the enzyme's catalytic activity, a delicate balance orchestrated by distant interactions. The communication between the allosteric and active sites unfolds through a network of connections of conformational changes, flexible regions, and specific amino acid residues. It is through this intricate web that the signal of change travels like a whisper carried on the wind. The protein's structure morphs, bending and shifting, as the signal journeys from the distant allosteric site to the bustling active site. Within this communication network lies a symphony of interacting residues, connected by the bonds of proximity or a series of intricate interactions. These interwoven pathways guide the transmission of the signal, allowing it to traverse the protein's intricate folds. It is a dance of hydrogen bonding, electrostatic interactions, and steric effects, each movement resonating through the intricate architecture. Certain amino acid residues step forward as key intermediaries in this grand communication. They possess unique roles, interacting directly with ligands or undergoing transformative changes that carry the signal forward. These exceptional residues, whether conserved or vital for structural integrity, hold the power to shape the enzyme's response to external cues. As the dance progresses, protein segments and domains emerge as conduits for the transmission of the signal. They possess unique features and dynamics, serving as bridges connecting functional sites or gracefully facilitating conformational changes. Like hinges in a grand door, they guide the motion, allowing the signal to flow seamlessly. The precise mechanism of signal transmission is a marvel, often a delicate interplay of conformational changes, communication networks, and the involvement of specific residues and segments. These mechanisms empower the enzyme to respond to its surroundings, regulating its activity and ensuring it performs its biological role with precision. In some enchanting cases, long-range communication unfolds through the subtle art of protein dynamics. Here, collective motions of domains or subunits facilitate the transfer of information across great distances within the enzyme's structure. It is a mesmerizing display, where the essence of the enzyme ripples through its very core. It's essential to appreciate that communication mechanisms can vary among different enzymes, with some showcasing shorter-range interactions and others embarking on long-range journeys. The precise details are dictated by the unique structure of each enzyme, the nature of its allosteric regulation, and the demands of its biological function. So, let us marvel at the enchanting communication of allosteric enzymes—a dance of distant domains and hidden pathways. Through conformational changes, communication networks, and the participation of extraordinary residues and segments, these enzymes respond to the whispers of their environment. They adapt, modulate their activity, and fulfill their intricate roles in the symphony of life.
Within the intricate world of enzymes, the existence and implementation of long-range communication mechanisms provide compelling evidence of intentional design. These mechanisms, observed in enzymes like allosteric enzymes, involve a captivating interplay of interconnected residues, specific amino acid interactions, and structural dynamics. Their precise arrangement and coordination hint at a level of complexity and precision that is often associated with intelligent design. Envision a network of intertwined elements working in unison, orchestrating the flow of information across vast distances within an enzyme. It is this intricate design that captivates scientists and prompts them to explore the origin and purpose behind these long-range communication pathways. Such pathways, carefully constructed, serve specific functional outcomes, leaving little room to chance. These communication mechanisms play a vital role in enzymes, serving as guardians of activity regulation and facilitators of coordination among multiple binding sites. The integration of these sites and the ability to transmit signals across considerable distances demand meticulous coordination and functional harmony. It is through this intricate orchestration that purposeful design reveals itself, for it is highly improbable for such mechanisms to arise randomly or through aimless processes. Imagine the transmission of signals, like whispers, traveling along specific amino acid residues, protein segments, and communication networks. It is through this intricate dance of information that these pathways come alive, rich with purpose. They bear the hallmarks of intelligent design, as they possess the ability to relay information to distant regions of the protein structure. The presence of pre-existing information, intricately encoded within the protein's blueprint, becomes evident as these signals navigate their predetermined routes. The beauty of long-range communication mechanisms in enzymes lies not only in their existence but also in their contribution to optimizing enzyme function. By modulating enzyme activity in an allosteric fashion, these mechanisms allow for fine-tuning and regulation of enzymatic processes. This remarkable ability to optimize function implies a deliberate design aimed at achieving specific objectives, driving efficiency and adaptability within the intricate machinery of life. As we delve into the depths of enzymes and their long-range communication, we uncover a symphony of purpose and intention. The interconnectedness of elements, the transmission of information, and the optimization of function all point toward intelligence at work—a guiding force behind the intricate design of these remarkable molecular machines. Join me as we continue to unravel the mysteries of life's intricacies, where fascination and scientific inquiry intertwine.
In the world of proteins, a phenomenon unfolds—structural plasticity. Like master artists, proteins possess the remarkable ability to assume different shapes and engage in dynamic fluctuations. And when a ligand enters the stage, the protein's dance takes on new dimensions. Ligand binding has the power to stabilize specific conformations or tip the delicate balance between different protein states. It is through this flexibility that proteins accommodate various ligands and orchestrate the transformative movements necessary for their grand performance. The conformational changes induced by ligand binding are no ordinary metamorphosis—they reflect shifts in the protein's energy landscape. Deep within the protein's core lie a multitude of conformational states, each with its own unique energy level. The entrance of a ligand changes the scene, favoring a new configuration—a ligand-bound conformation with lower energy. The protein transitions gracefully, embracing this new state, leaving behind the ensemble of possibilities that once adorned its stage. To capture the intricate choreography of ligand-induced conformational changes is no simple feat. Experimental techniques like X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) offer glimpses into these structural metamorphoses. Yet, to unveil the full breadth of the protein's dance requires a symphony of computational modeling techniques. It is an art of precision, where every movement must be orchestrated flawlessly to achieve the desired functional outcome. These conformational changes are not merely for show—they optimize binding affinity, selectivity, and catalytic activity. Each movement aligns the residues within the binding pocket with impeccable precision, ensuring efficient ligand recognition and enzymatic prowess. The complexity lies in the intricate interplay of factors—the inherent flexibility of proteins, the energetic considerations, and the functional demands. The protein's performance is a delicate balance, a harmonious fusion of all these elements. And amidst this dance, water molecules take center stage. They, too, play a crucial role in mediating the interactions between the protein and its ligand. With their nimble nature, they form delicate hydrogen bonds, joining both parties in an elegant embrace. These water molecules stabilize the binding interaction, adding to the overall affinity and contributing to recognition. It is this interplay—the fluidity of proteins, the energy landscapes, the functional intricacies—that ensures the exquisite placement of residues within the binding pocket. Each movement of the dance enables specific and efficient interactions, as proteins and ligands find their destined embrace. It is a dance of specificity, a waltz of elegance observed in the wonders of the natural world. Each shift in shape brings forth a new functional landscape, optimizing the protein's performance. And amidst it all, water molecules join the symphony, delicately stabilizing the bonds that unite protein and ligand. It is through this intricate interplay that the beauty of biological specificity unfolds before our eyes.
In the intricate dance of enzymatic reactions, a remarkable event unfolds as the enzyme carefully positions the phosphate group of GTP near the 6th carbon atom of IMP—a critical moment in the synthesis of adenylosuccinate and the subsequent formation of AMP. As the stage is set, a remarkable transformation takes place through a nucleophilic attack, a moment of molecular connection and transformation. Like a skilled performer, the phosphate group from GTP launches an energetic assault on the 6th carbon atom of IMP—a nucleophilic attack. This decisive move triggers a cascade of events, as the phosphate group is transferred from the terminal position of GTP to the waiting 6th carbon atom of IMP. This transfer, this phosphorylation, heralds the birth of adenylosuccinate 6-phosphate—an essential step on the path to the formation of adenylosuccinate itself. This enzymatic symphony is made possible by the enzyme's masterful guidance and the provision of an appropriate environment—a specialized active site that sets the stage for this chemical union. Within this carefully crafted environment, the transfer of the phosphate group becomes possible, as the enzyme's structure aligns the reacting molecules in perfect harmony. It is within this orchestrated setting that the magic of phosphorylation occurs, a key step that breathes life into the formation of adenylosuccinate. It is crucial to note that adenylosuccinate synthase, the conductor of this symphony, also oversees other essential steps in this grand production. As described previously, these steps involve the binding of additional substrates, the cleavage of GTP, the activation of aspartate, and the elegant condensation of activated aspartate with adenylosuccinate 6-phosphate. Together, these intricate movements lead to the ultimate creation of adenylosuccinate—a vital intermediate that paves the way for the biosynthesis of AMP, a molecule of paramount importance in cellular processes. In this captivating journey of molecular transformations, we witness the delicate interplay between enzymes and their substrates. The transfer of the phosphate group, orchestrated by adenylosuccinate synthase, stands as a pivotal moment, ushering us closer to the formation of AMP. Join me as we continue to explore the wonders of biochemistry, where these intricate dances of molecules shape the very essence of life itself.
Step 2: Cleavage of GTP
In the second step, the GTP molecule that donated the phosphate group in the previous step undergoes cleavage. The cleavage of GTP yields guanosine diphosphate (GDP) and inorganic phosphate (Pi). This step is essential for the overall reaction and allows for the recycling of GDP for future reactions.
Step 3: Aspartate Activation
Next, the enzyme catalyzes the activation of aspartate by adding a phosphate group to it. This step requires ATP as a cofactor. The phosphate group is transferred from ATP to aspartate, resulting in the formation of phosphoribosylaminoimidazole carboxylate (AICAR) and pyrophosphate (PPi).
Step 4: Formation of Adenylosuccinate
In the final step, the activated aspartate (AICAR) is condensed with adenylosuccinate 6-phosphate produced in the first step. This condensation reaction leads to the formation of adenylosuccinate. The enzyme catalyzes the formation of a carbon-nitrogen bond between the carboxyl group of aspartate and the amino group of adenylosuccinate 6-phosphate.
Within the intricate unfolding of biochemical reactions, adenylosuccinate synthase takes the stage, performing a mesmerizing multistep routine. This enzyme orchestrates a symphony of phosphorylation, cleavage, activation, and condensation, all in pursuit of the formation of adenylosuccinate—the vital intermediary in AMP biosynthesis. With graceful precision, adenylosuccinate synthase facilitates the transfer of phosphate groups and the delicate formation of carbon-nitrogen bonds, contributing to the creation of essential nucleotides within the cell. Yet, adenylosuccinate synthase is not merely a solo performer—it operates within a complex regulatory framework. Feedback inhibition by AMP and other nucleotides ensures a delicate balance of nucleotide levels, maintaining harmony within the cell. The enzyme's activity can be further modulated through gene expression regulation and post-translational modifications, adding layers of control to its performance. In the intricate web of factors influencing adenylosuccinate synthase's activity, substrate availability, the presence of vital cofactors like Mg2+, and regulatory mechanisms dance together. Each step in the enzyme's routine is finely tuned to ensure its proper function. Any modifications or mutations that affect its structure or function can sway the rhythm of its performance. The significance of adenylosuccinate synthase stretches beyond its catalytic prowess and substrate specificity. It plays a fundamental role in nucleotide biosynthesis, a vital process for cellular metabolism and genetic processes. Its presence across diverse organisms speaks to its evolutionary conservation and its importance in sustaining the delicate balance of life. But when we delve into the origins of adenylosuccinate synthase, we encounter challenges that beckon us to contemplate. This intricate enzyme with its precise amino acid sequences, folding into a three-dimensional structure, hints at a grand design rather than blind chemical processes. The remarkable complexity and specific interactions necessary for its activity suggest the involvement of an intelligent agent. Enzymes, like adenylosuccinate synthase, bear the signature of irreducible complexity. They require multiple interacting components to function harmoniously. Gradual step-by-step processes struggle to account for the simultaneous appearance of all the necessary components. The intricate shape, active site, and catalytic properties of enzymes hint at intentional design rather than random chance. We must consider the limitations of mere chemical processes when unraveling the origins of such sophisticated biochemical systems. The probability of enzymes like adenylosuccinate synthase emerging through unguided events is staggeringly low. Given the intricate functional requirements and complex interactions involved, an intelligent designer offers a more plausible explanation for their origins. It is within the embrace of this perspective that we can appreciate the wonder and awe-inspiring complexity of adenylosuccinate synthase and other intricate biochemical systems. Adenylosuccinate synthase's choreography of phosphorylation, cleavage, activation, and condensation is a performance that orchestrates the synthesis of crucial nucleotides, while the ensemble of regulatory mechanisms ensures cellular harmony. And as we explore its origins, let us ponder the delicate balance between blind chemistry and the fingerprints of an intelligent designer.
Adenylosuccinate lyase (ASL)
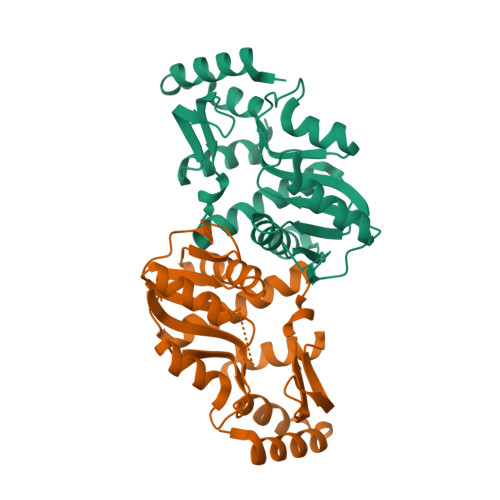
In the intricate realm of cellular chemistry, Adenylosuccinate lyase (ASL) emerges as a remarkable performer. This enzyme takes on the task of cleaving adenylosuccinate—a crucial intermediate molecule—into AMP (adenosine monophosphate) and fumarate. This elegant reaction unfolds within the cytoplasm, marking the second step in the de novo synthesis of AMP. ASL's delicate choreography involves severing the bond between adenosine and succinate within adenylosuccinate. With this precise maneuver, AMP is born, poised to play its essential role in a myriad of cellular processes, from energy metabolism to RNA synthesis and signaling pathways. ASL, typically existing as a solitary entity, consists of a single polypeptide chain. Its structure embraces the captivating TIM barrel fold, a widely embraced motif in the world of enzymes. Named after the triosephosphate isomerase, the TIM barrel fold showcases an enchanting arrangement of alpha helices and beta strands, forming a barrel-like structure. This exquisite fold often boasts eight parallel strands of beta sheet interwoven with alpha helices, creating a rhythmic alternation around the core axis. The TIM barrel fold follows a choreographed pattern—strands adjacent, hands linked in loops or short helices. The grand finale of this structural symphony unfolds as the N- and C-termini of the protein unite, completing the barrel's poetic embrace. The interior of the TIM barrel, a sacred space, holds the enzyme's active site, rich in hydrophobic contours. This sanctum welcomes specific substrates and ligands, forming intimate interactions and enabling the enzyme's catalytic magic. The TIM barrel fold, with its versatile nature, captivates the scientific stage. Its diverse repertoire extends to enzymes performing vital roles in catalysis, isomerization, and metabolic pathways. This fold provides a stable and rigid framework, an unyielding canvas for the artist of catalysis to work their wonders. Its dance within the TIM barrel fold, a feat of structure and function, illuminates the intricate beauty of enzymes. In the grand theater of cellular chemistry, ASL takes center stage, painting a vivid picture of the wonders of life's molecular choreography.
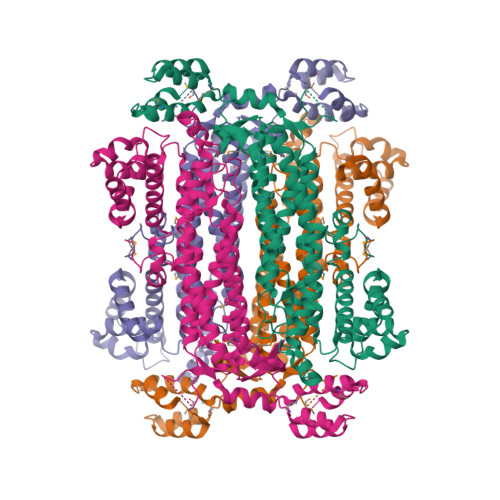
Within the heart of Adenylosuccinate lyase (ASL), an active site brims with captivating activity. This enzyme possesses a unique talent—it acts specifically on adenylosuccinate, an exquisite substrate, cleaving it with precision to unveil the treasures within: AMP and fumarate. The active site of ASL serves as a stage for molecular interactions, where specific amino acid residues choreograph the enzyme's performance. Their strategic placement within the active site ensures the seamless execution of the enzymatic reaction, like a well-rehearsed dance. ASL's grand performance can be orchestrated by factors beyond its stage. At the transcriptional level, the expression of the gene responsible for producing this marvelous enzyme can be influenced by the cellular environment. The availability of purine nucleotides, those precious building blocks of life, can sway the expression of ASL and set the stage for its enzymatic prowess. Additionally, post-translational modifications and intricate interactions with other proteins can add nuances to ASL's performance, enhancing or modulating its activity. As a solitary performer, ASL thrives in its monomeric form. Its amino acid sequence, a blueprint etched in the language of life, ensures that the enzyme is poised for optimal function. Every letter in this sequence holds significance, contributing to the structural arrangement that allows ASL to unleash its catalytic powers with unrivaled efficiency. So let us marvel at the grand performance of Adenylosuccinate lyase, as it takes center stage, delicately binding to its adenylosuccinate partner and gracefully cleaving it to reveal the hidden treasures of AMP and fumarate. The expression of this captivating enzyme is finely tuned, responding to the needs of the cellular orchestra. In its monomeric glory, ASL dances through the intricacies of the enzymatic world, leaving us in awe of the harmonious interplay of molecules that underpins the symphony of life.
In the grand saga of nucleotide synthesis, IMP takes center stage as a precursor for two vital nucleotides—AMP and GMP. These molecular heroes, AMP and GMP, hold key roles in living systems. Let's unravel their secrets and discover the magic they bring to the stage. AMP, also known as adenosine monophosphate, is a nucleotide of great significance. Its structure is a trio of marvels—a nitrogenous base named adenine, a sugar known as ribose, and a single phosphate group. Together, they form the foundation of AMP's power. This nucleotide luminary plays an essential role in a multitude of biological processes, orchestrating cellular activities with grace and precision. Now, let's shift our gaze to GMP—guanosine monophosphate—an esteemed member of the nucleotide family. Its structure closely resembles that of AMP, with a nitrogenous base called guanine, a ribose sugar, and a single phosphate group. GMP, like its counterpart AMP, holds a pivotal position in the intricate realm of nucleic acids. It contributes to the formation of DNA and RNA, participating in the dance of life's information storage and transmission. Why were AMP and GMP chosen to be part of the extraordinary quartet of RNA and DNA? The answer lies in their structural and functional prowess. Alongside their faithful partners, T and C, they contribute to the rich diversity of base pairings, elevating the informational content of DNA and RNA to new heights. The presence of AMP and GMP in this quartet is not a coincidence but a deliberate selection. Nature, in its wisdom, has chosen these nucleotides for their ability to form stable base pairs and their seamless integration within the grand structure and function of DNA and RNA. It is through the exquisite arrangement and interplay of these nucleotides that the quartet—A, T/U, G, and C—unleashes the incredible genetic diversity and capacity for storing information witnessed in all living organisms. So, let us marvel at the beauty of AMP and GMP as they join their counterparts, T and C, in the grand tale of nucleotide synthesis. Their unique structures and roles have been carefully curated to ensure the symphony of life plays out with precision. Together, they contribute to the richness of genetic information, encapsulating the wonder and complexity of the living world.
AMP synthesis
The journey from IMP to AMP and GMP unfolds through distinct pathways, each with its own set of pivotal reactions. Let's embark on this captivating adventure and explore the remarkable transformations that take place. In the realm of AMP synthesis, the first step is orchestrated by the masterful enzyme adenylosuccinate synthase, also known as adenylosuccinase. This enzymatic maestro guides the transfer of the amino group from aspartate to IMP, forging a bond between aspartate and IMP. This reaction is fueled by the energetic dance of GTP, as it sacrifices itself through hydrolysis. GTP, a nucleotide composed of a guanine base, a ribose sugar, and three phosphate groups, is a true cellular powerhouse. It serves as an energy carrier, fueling diverse processes such as protein synthesis, signal transduction, and energy metabolism. When GTP undergoes hydrolysis, a water molecule steps onto the stage, leading to the breaking of a high-energy phosphate bond. The bond between the last two phosphate groups in GTP is cleaved, resulting in the birth of GDP and the release of inorganic phosphate (Pi). GDP, formed by the removal of a phosphate group from GTP, emerges as a transformed molecule, while Pi dances its way as a separate entity. Enzymes called GTPases are the skilled facilitators of this hydrolysis dance, playing essential roles in cellular processes like G protein signaling and protein synthesis regulation. As GTP bids adieu, it leaves behind a trail of released energy, a gift for the cell to utilize in various energy-demanding endeavors. Now, armed with adenylosuccinate, our journey ventures further into the second crucial reaction. Here, adenylosuccinate lyase takes the spotlight, gracefully removing the fumarate group from adenylosuccinate. As this transformative enzyme wields its power, a wondrous alchemy occurs—AMP is born. It's important to note that adenylosuccinate lyase also catalyzes Reaction 9 of the IMP pathway, where it removes fumarate from another intermediate. Both reactions share a common thread—they add a nitrogen atom to the molecule, with fumarate emerging as a byproduct, bidding farewell to the stage. In this enthralling tale of nucleotide synthesis, we witness the magic of transformation. AMP, once a distant dream in the realm of IMP, takes shape through the orchestrated dances of adenylosuccinate synthase and adenylosuccinate lyase. The vibrant energy of GTP fuels these reactions, guiding the steps of molecular ballet. And as fumarate bids its farewell, AMP emerges as a triumphant hero, ready to fulfill its role in the grand symphony of DNA and RNA. So, let us marvel at the wonders of these reactions as we journey from IMP to AMP and GMP. It is a dance of molecular elegance, where bonds are formed, hydrolysis unlocks energy, and transformation brings forth new life. Through these remarkable processes, the intricate world of nucleotide synthesis unveils its captivating secrets, revealing the exquisite mechanisms that underpin the dance of life.
GMP synthesis
But wait, the path to GMP from IMP takes a different course—a fascinating journey that unfolds through a separate pathway. Let's embark on this alternate adventure and witness the captivating transformations that occur along the way. Our tale begins with the dehydrogenation of IMP, a pivotal moment orchestrated by the enzyme IMP dehydrogenase. This enzymatic marvel guides the removal of hydrogen from IMP, resulting in the reduction of NAD+ and the birth of xanthosine monophosphate (XMP). XMP, the ribonucleotide of the base xanthine, emerges as a new character in our narrative. This reaction holds a subtle dance of electrons, as IMP surrenders its hydrogen atoms, leading to the transformation of NAD+. Through this exchange, XMP takes its rightful place, ready to continue the journey. Now, with XMP at the center stage, the story progresses to its climax. Here, the enzyme GMP synthetase takes the spotlight, orchestrating the transfer of the amide nitrogen from glutamine to XMP. This transformative moment marks the birth of GMP. But the magic doesn't stop there—the energy required for this transfer reaction comes from the hydrolysis of ATP. ATP, the universal energy currency of cells, undergoes a mesmerizing transformation, splitting into AMP and pyrophosphate (PPi). It is through this captivating hydrolysis that the necessary energy is harnessed to drive the transfer of the amide nitrogen from glutamine to XMP. As the ATP molecule sacrifices itself, AMP emerges, and pyrophosphate is released as a separate entity. And there it is—GMP, the result of this enchanting sequence of events. Now, armed with its new identity, GMP stands ready to fulfill its role in cellular processes. It becomes a player in the grand symphony of life, contributing to the dance of DNA and RNA, fueling the intricate mechanisms that drive the wonders of biology. In this captivating narrative of nucleotide synthesis, we witness the unique pathway from IMP to GMP. IMP dehydrogenase sets the stage with its dehydrogenation prowess, giving rise to XMP. Then, GMP synthetase takes command, conducting the transfer of the amide nitrogen from glutamine to XMP, with the energetic support of ATP hydrolysis. It is through these alchemical processes that GMP is born, ready to embark on its role in the cellular symphony. So, let us rejoice in the wonders of this journey—a path that diverges from AMP synthesis to lead us to the realm of GMP. It is a dance of molecular transformations, where bonds are forged, energy is harnessed, and new characters step onto the stage. Through these magnificent processes, the world of nucleotide synthesis reveals its secrets, unraveling the intricate mechanisms that underlie the beauty of life's dance.

Adenylosuccinate synthase (ADSS)
Adenylosuccinate synthase (ADSS) is a remarkable enzyme responsible for catalyzing the formation of adenylosuccinate—a vital intermediate in the synthesis of adenosine monophosphate (AMP). AMP, in turn, plays a pivotal role in numerous cellular processes, underscoring the significance of ADSS in the intricate machinery of life. Let us embark on a journey to explore the intricacies of this enzyme and the fascinating mechanisms by which it acquires the essential cofactors for its catalytic prowess. ADSS, with a total structure weight of 48.27 kDa (kilodaltons) and an awe-inspiring assembly of 3,553 atoms, stands as a testament to the intricacy and precision of biological machinery. Its delicate balance of atoms and subunits enables it to orchestrate the formation of adenylosuccinate—a vital stepping stone on the path to AMP. To unlock its catalytic potential, ADSS relies on the presence of divalent metal ions, particularly the versatile magnesium (Mg2+). These ions act as cofactors, deftly facilitating the enzyme's catalytic activities. But how does the simplest of cells acquire these precious metal ions? The acquisition of divalent metal ions, such as magnesium, can occur through various mechanisms. One such mechanism is passive diffusion—an elegant dance between the ions and the cell membrane. Picture a scenario where the concentration of these ions is higher outside the cell compared to within its cytoplasm. In this setting, the ions possess a powerful driving force, compelling them to traverse the cell membrane and journey into the cell's inner sanctum. Through passive diffusion, these divalent metal ions, including the esteemed magnesium, spontaneously traverse the cell membrane, propelled by the concentration gradient. They gracefully move from regions of higher concentration—found outside the cell—to areas of lower concentration nestled within its delicate confines. This captivating dance continues until a harmonious equilibrium is reached, where the concentration of these ions is equal both inside and outside the cell. What distinguishes passive diffusion is its ability to transport small, uncharged molecules or ions that can effortlessly navigate the lipid bilayer of the cell membrane. These agile particles effortlessly slip through the molecular gaps, driven solely by the forces of chance and concentration. Remarkably, this process demands no additional energy or the involvement of specialized transport proteins, allowing the ions to traverse freely. However, it is crucial to note that the elegance of passive diffusion is limited to these small, uncharged entities. Larger molecules or charged ions, with their bulk and electric charges, require more intricate means of transportation. For their triumphant passage across the cell membrane, specific channels, transporters, or pumps must come into play, guiding these larger and charged entities on their destined path. As we peel back the layers of ADSS and its quest for magnesium ions, we uncover a tale of delicate balance and captivating dance. The enzyme's role in the production of AMP, intertwined with the acquisition of essential cofactors, paints a vivid picture of the marvels hidden within the microscopic world of cells. Join me as we continue to explore the remarkable intricacies of the molecular realm, where beauty and scientific wonder coexist in perfect harmony.
In the realm of simple cells like bacteria or archaea, fascinating concentration differences take place, specifically with divalent metal ions such as magnesium (Mg2+). Let's venture into this world and explore the mechanisms that establish these differences, painting a vivid picture of cellular life. One source of concentration disparity arises from the contrasting Mg2+ ion levels between the cell's external environment and its cytoplasm. Picture a cell residing in a surrounding medium with higher concentrations of Mg2+ ions than those found within its cytoplasm. This natural variance sets the stage for a wondrous play of ion dynamics. But there's more to this tale. Simple cells possess metabolic processes that actively manipulate Mg2+ ions within their cytoplasm. Enzymes and transporters engaged in cellular metabolism rely on Mg2+ ions as cofactors, consuming them in their biochemical endeavors. Through this consumption, concentration gradients are established, further enhancing the contrast between the extracellular environment and the cell's inner sanctum. Transport systems within these cells play a key role in sculpting the concentration differences. Imagine ion channels or transporters adorning the cell membrane, acting as selective gatekeepers. They facilitate the movement of Mg2+ ions, actively transporting them into the cell or ushering them out. This dynamic interplay ensures that concentration differences persist, adding a thrilling dimension to the cellular landscape. Ah, but we must not forget the cell's membrane potential, a conductor orchestrating this ion symphony. The membrane potential, the difference in electrical charge across the cell membrane, exerts its influence on charged ions like Mg2+. This electrical potential alters the ions' movement, creating disparities between the extracellular environment and the cytoplasm. It's a delicate interplay of charges, intricately shaping the ion concentrations within the cell. In the realm of cellular homeostasis, regulatory mechanisms come into play, maintaining the delicate balance of ion concentrations, including the precious Mg2+. Ion channels, transporters, and regulatory proteins take center stage, masterfully allowing or blocking the passage of ions across the membrane. With their guidance, concentration differences persist, ensuring that the dance of ions continues harmoniously within the cell. So, let us revel in the enchantment of concentration differences in simple cells, where Mg2+ ions dictate the ebb and flow of cellular life. Through mechanisms of active accumulation, consumption, and transport, these cells establish captivating disparities. And with the aid of membrane potentials and regulatory proteins, the delicate balance of ion concentrations is preserved, ensuring the dance of life remains in perfect harmony.
Step 1: Phosphorylation of IMP
Within the realm of enzymes, there lies a captivating character known as adenylosuccinate synthase. This remarkable enzyme belongs to the esteemed class of ligases, specifically the family of transferases that are skilled in forming carbon-nitrogen bonds. Let us embark on a journey through the multistep dance orchestrated by this enchanting enzyme, as it weaves together substrates and brings forth the birth of adenylosuccinate. Our tale begins with the union of two key players: inosine monophosphate (IMP) and aspartate. Picture these molecules intertwining, their atoms aligning with precision, ready to embark on a transformation of extraordinary significance. The adenylosuccinate synthase, with its enzymatic mastery, guides the way, catalyzing a symphony of chemical reactions. Step by step, the dance unfolds. In the first movement, IMP and aspartate come together, forming an intermediate known as adenylosuccinyl-AMP (ASAMP). This intermediate bears the potential for greatness, harboring the essence of adenylosuccinate within its intricate structure. But the transformation doesn't end there. The dance continues, and with each step, the ASAMP intermediate undergoes a graceful metamorphosis. In the second movement, a remarkable rearrangement occurs, leading to the formation of an energy-rich molecule called adenosine monophosphate (AMP). This pivotal step is a crucial milestone in the de novo synthesis of AMP, a molecule of great significance in the cellular symphony of life. As we witness the unfolding of this multistep reaction, we are captivated by the precision and elegance with which adenylosuccinate synthase orchestrates the union of IMP and aspartate. Its skilled hand guides the formation of adenylosuccinate, paving the way for the subsequent formation of AMP. Each step in this enchanting dance serves a purpose, bringing us closer to the grand finale. So, let us revel in the magic of adenylosuccinate synthase, a ligase of exceptional talent. It masterfully catalyzes the formation of adenylosuccinate through a captivating multistep reaction. The fusion of IMP and aspartate unveils the hidden potential within, creating a symphony of molecules that sets the stage for the birth of AMP. It is through the expertise of this remarkable enzyme that the de novo synthesis of AMP finds its path, contributing to the harmonious dance of cellular life.
Implementing conformational change functions in the protein
In the intricate dance of molecular interactions, conformational changes hold the power to elevate binding interactions to new heights. When a ligand or substrate embraces the binding pocket of a protein, a symphony of transformations is set into motion. These changes add a touch of complexity, captivating our senses and enriching the process with a mesmerizing allure. As the ligand finds its place within the binding pocket, it whispers secrets to the protein, provoking a response. The protein, like a maestro, orchestrates its movements, shifting and rearranging to accommodate the presence of its partner. Individual amino acid residues may gracefully adjust their positions, delicately harmonizing with the ligand. In some cases, larger-scale transformations occur, where entire protein domains gracefully shift, adapting to the newfound connection. These conformational changes are born from the intimate dialogue between the ligand and specific residues within the binding pocket. Like a dance partner guiding their companion, the ligand influences the protein's structure, imbuing it with a fresh elegance. But the impact doesn't end there—these changes can ripple through the protein, transcending the immediate vicinity of the binding site. This captivating phenomenon is known as allosteric regulation, where the binding event resonates with distant regions of the protein. Through allosteric effects, the protein's activity, stability, and interactions with other molecules are transformed beyond the confines of the binding site. It's as if the protein, awakened by the ligand's touch, communicates its newfound knowledge to its surroundings. This communication between the binding site and other regions of the protein serves as a vital mechanism for regulating cellular processes, a symphony of coordination in the intricate dance of life. So, let us marvel at the wonders of conformational changes induced by ligand binding. They enrich the binding interaction, enhancing specificity and affinity. As the ligand whispers its secrets, the protein responds, reshaping itself with grace and elegance. This transformative dance captivates our imagination, as allosteric effects ripple through the protein, influencing its activity, stability, and interactions. It is through this intricate communication that the protein navigates the complex symphony of cellular processes, unveiling the grandeur of nature's choreography.
Long distant signaling through allosteric networks points to a designed setup
In the amazing world of enzymes, we encounter a remarkable phenomenon known as allosteric regulation. Within these enzymes, a symphony of communication unfolds, spanning vast distances. Imagine an intricate dance between distinct binding sites—the active site, where the substrate finds its place, and the allosteric site, a distant domain with a secret to tell. This interplay of binding events holds the key to modulating the enzyme's activity, and one fascinating example is the renowned enzyme adenylosuccinate synthase. As adenylosuccinate synthase enters the spotlight, we witness its transformation through conformational changes triggered by ligand binding at the allosteric site. This intricate ballet of motion governs the enzyme's catalytic activity, a delicate balance orchestrated by distant interactions. The communication between the allosteric and active sites unfolds through a network of connections of conformational changes, flexible regions, and specific amino acid residues. It is through this intricate web that the signal of change travels like a whisper carried on the wind. The protein's structure morphs, bending and shifting, as the signal journeys from the distant allosteric site to the bustling active site. Within this communication network lies a symphony of interacting residues, connected by the bonds of proximity or a series of intricate interactions. These interwoven pathways guide the transmission of the signal, allowing it to traverse the protein's intricate folds. It is a dance of hydrogen bonding, electrostatic interactions, and steric effects, each movement resonating through the intricate architecture. Certain amino acid residues step forward as key intermediaries in this grand communication. They possess unique roles, interacting directly with ligands or undergoing transformative changes that carry the signal forward. These exceptional residues, whether conserved or vital for structural integrity, hold the power to shape the enzyme's response to external cues. As the dance progresses, protein segments and domains emerge as conduits for the transmission of the signal. They possess unique features and dynamics, serving as bridges connecting functional sites or gracefully facilitating conformational changes. Like hinges in a grand door, they guide the motion, allowing the signal to flow seamlessly. The precise mechanism of signal transmission is a marvel, often a delicate interplay of conformational changes, communication networks, and the involvement of specific residues and segments. These mechanisms empower the enzyme to respond to its surroundings, regulating its activity and ensuring it performs its biological role with precision. In some enchanting cases, long-range communication unfolds through the subtle art of protein dynamics. Here, collective motions of domains or subunits facilitate the transfer of information across great distances within the enzyme's structure. It is a mesmerizing display, where the essence of the enzyme ripples through its very core. It's essential to appreciate that communication mechanisms can vary among different enzymes, with some showcasing shorter-range interactions and others embarking on long-range journeys. The precise details are dictated by the unique structure of each enzyme, the nature of its allosteric regulation, and the demands of its biological function. So, let us marvel at the enchanting communication of allosteric enzymes—a dance of distant domains and hidden pathways. Through conformational changes, communication networks, and the participation of extraordinary residues and segments, these enzymes respond to the whispers of their environment. They adapt, modulate their activity, and fulfill their intricate roles in the symphony of life.
Within the intricate world of enzymes, the existence and implementation of long-range communication mechanisms provide compelling evidence of intentional design. These mechanisms, observed in enzymes like allosteric enzymes, involve a captivating interplay of interconnected residues, specific amino acid interactions, and structural dynamics. Their precise arrangement and coordination hint at a level of complexity and precision that is often associated with intelligent design. Envision a network of intertwined elements working in unison, orchestrating the flow of information across vast distances within an enzyme. It is this intricate design that captivates scientists and prompts them to explore the origin and purpose behind these long-range communication pathways. Such pathways, carefully constructed, serve specific functional outcomes, leaving little room to chance. These communication mechanisms play a vital role in enzymes, serving as guardians of activity regulation and facilitators of coordination among multiple binding sites. The integration of these sites and the ability to transmit signals across considerable distances demand meticulous coordination and functional harmony. It is through this intricate orchestration that purposeful design reveals itself, for it is highly improbable for such mechanisms to arise randomly or through aimless processes. Imagine the transmission of signals, like whispers, traveling along specific amino acid residues, protein segments, and communication networks. It is through this intricate dance of information that these pathways come alive, rich with purpose. They bear the hallmarks of intelligent design, as they possess the ability to relay information to distant regions of the protein structure. The presence of pre-existing information, intricately encoded within the protein's blueprint, becomes evident as these signals navigate their predetermined routes. The beauty of long-range communication mechanisms in enzymes lies not only in their existence but also in their contribution to optimizing enzyme function. By modulating enzyme activity in an allosteric fashion, these mechanisms allow for fine-tuning and regulation of enzymatic processes. This remarkable ability to optimize function implies a deliberate design aimed at achieving specific objectives, driving efficiency and adaptability within the intricate machinery of life. As we delve into the depths of enzymes and their long-range communication, we uncover a symphony of purpose and intention. The interconnectedness of elements, the transmission of information, and the optimization of function all point toward intelligence at work—a guiding force behind the intricate design of these remarkable molecular machines. Join me as we continue to unravel the mysteries of life's intricacies, where fascination and scientific inquiry intertwine.
In the world of proteins, a phenomenon unfolds—structural plasticity. Like master artists, proteins possess the remarkable ability to assume different shapes and engage in dynamic fluctuations. And when a ligand enters the stage, the protein's dance takes on new dimensions. Ligand binding has the power to stabilize specific conformations or tip the delicate balance between different protein states. It is through this flexibility that proteins accommodate various ligands and orchestrate the transformative movements necessary for their grand performance. The conformational changes induced by ligand binding are no ordinary metamorphosis—they reflect shifts in the protein's energy landscape. Deep within the protein's core lie a multitude of conformational states, each with its own unique energy level. The entrance of a ligand changes the scene, favoring a new configuration—a ligand-bound conformation with lower energy. The protein transitions gracefully, embracing this new state, leaving behind the ensemble of possibilities that once adorned its stage. To capture the intricate choreography of ligand-induced conformational changes is no simple feat. Experimental techniques like X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) offer glimpses into these structural metamorphoses. Yet, to unveil the full breadth of the protein's dance requires a symphony of computational modeling techniques. It is an art of precision, where every movement must be orchestrated flawlessly to achieve the desired functional outcome. These conformational changes are not merely for show—they optimize binding affinity, selectivity, and catalytic activity. Each movement aligns the residues within the binding pocket with impeccable precision, ensuring efficient ligand recognition and enzymatic prowess. The complexity lies in the intricate interplay of factors—the inherent flexibility of proteins, the energetic considerations, and the functional demands. The protein's performance is a delicate balance, a harmonious fusion of all these elements. And amidst this dance, water molecules take center stage. They, too, play a crucial role in mediating the interactions between the protein and its ligand. With their nimble nature, they form delicate hydrogen bonds, joining both parties in an elegant embrace. These water molecules stabilize the binding interaction, adding to the overall affinity and contributing to recognition. It is this interplay—the fluidity of proteins, the energy landscapes, the functional intricacies—that ensures the exquisite placement of residues within the binding pocket. Each movement of the dance enables specific and efficient interactions, as proteins and ligands find their destined embrace. It is a dance of specificity, a waltz of elegance observed in the wonders of the natural world. Each shift in shape brings forth a new functional landscape, optimizing the protein's performance. And amidst it all, water molecules join the symphony, delicately stabilizing the bonds that unite protein and ligand. It is through this intricate interplay that the beauty of biological specificity unfolds before our eyes.
In the intricate dance of enzymatic reactions, a remarkable event unfolds as the enzyme carefully positions the phosphate group of GTP near the 6th carbon atom of IMP—a critical moment in the synthesis of adenylosuccinate and the subsequent formation of AMP. As the stage is set, a remarkable transformation takes place through a nucleophilic attack, a moment of molecular connection and transformation. Like a skilled performer, the phosphate group from GTP launches an energetic assault on the 6th carbon atom of IMP—a nucleophilic attack. This decisive move triggers a cascade of events, as the phosphate group is transferred from the terminal position of GTP to the waiting 6th carbon atom of IMP. This transfer, this phosphorylation, heralds the birth of adenylosuccinate 6-phosphate—an essential step on the path to the formation of adenylosuccinate itself. This enzymatic symphony is made possible by the enzyme's masterful guidance and the provision of an appropriate environment—a specialized active site that sets the stage for this chemical union. Within this carefully crafted environment, the transfer of the phosphate group becomes possible, as the enzyme's structure aligns the reacting molecules in perfect harmony. It is within this orchestrated setting that the magic of phosphorylation occurs, a key step that breathes life into the formation of adenylosuccinate. It is crucial to note that adenylosuccinate synthase, the conductor of this symphony, also oversees other essential steps in this grand production. As described previously, these steps involve the binding of additional substrates, the cleavage of GTP, the activation of aspartate, and the elegant condensation of activated aspartate with adenylosuccinate 6-phosphate. Together, these intricate movements lead to the ultimate creation of adenylosuccinate—a vital intermediate that paves the way for the biosynthesis of AMP, a molecule of paramount importance in cellular processes. In this captivating journey of molecular transformations, we witness the delicate interplay between enzymes and their substrates. The transfer of the phosphate group, orchestrated by adenylosuccinate synthase, stands as a pivotal moment, ushering us closer to the formation of AMP. Join me as we continue to explore the wonders of biochemistry, where these intricate dances of molecules shape the very essence of life itself.
Step 2: Cleavage of GTP
In the second step, the GTP molecule that donated the phosphate group in the previous step undergoes cleavage. The cleavage of GTP yields guanosine diphosphate (GDP) and inorganic phosphate (Pi). This step is essential for the overall reaction and allows for the recycling of GDP for future reactions.
Step 3: Aspartate Activation
Next, the enzyme catalyzes the activation of aspartate by adding a phosphate group to it. This step requires ATP as a cofactor. The phosphate group is transferred from ATP to aspartate, resulting in the formation of phosphoribosylaminoimidazole carboxylate (AICAR) and pyrophosphate (PPi).
Step 4: Formation of Adenylosuccinate
In the final step, the activated aspartate (AICAR) is condensed with adenylosuccinate 6-phosphate produced in the first step. This condensation reaction leads to the formation of adenylosuccinate. The enzyme catalyzes the formation of a carbon-nitrogen bond between the carboxyl group of aspartate and the amino group of adenylosuccinate 6-phosphate.
Within the intricate unfolding of biochemical reactions, adenylosuccinate synthase takes the stage, performing a mesmerizing multistep routine. This enzyme orchestrates a symphony of phosphorylation, cleavage, activation, and condensation, all in pursuit of the formation of adenylosuccinate—the vital intermediary in AMP biosynthesis. With graceful precision, adenylosuccinate synthase facilitates the transfer of phosphate groups and the delicate formation of carbon-nitrogen bonds, contributing to the creation of essential nucleotides within the cell. Yet, adenylosuccinate synthase is not merely a solo performer—it operates within a complex regulatory framework. Feedback inhibition by AMP and other nucleotides ensures a delicate balance of nucleotide levels, maintaining harmony within the cell. The enzyme's activity can be further modulated through gene expression regulation and post-translational modifications, adding layers of control to its performance. In the intricate web of factors influencing adenylosuccinate synthase's activity, substrate availability, the presence of vital cofactors like Mg2+, and regulatory mechanisms dance together. Each step in the enzyme's routine is finely tuned to ensure its proper function. Any modifications or mutations that affect its structure or function can sway the rhythm of its performance. The significance of adenylosuccinate synthase stretches beyond its catalytic prowess and substrate specificity. It plays a fundamental role in nucleotide biosynthesis, a vital process for cellular metabolism and genetic processes. Its presence across diverse organisms speaks to its evolutionary conservation and its importance in sustaining the delicate balance of life. But when we delve into the origins of adenylosuccinate synthase, we encounter challenges that beckon us to contemplate. This intricate enzyme with its precise amino acid sequences, folding into a three-dimensional structure, hints at a grand design rather than blind chemical processes. The remarkable complexity and specific interactions necessary for its activity suggest the involvement of an intelligent agent. Enzymes, like adenylosuccinate synthase, bear the signature of irreducible complexity. They require multiple interacting components to function harmoniously. Gradual step-by-step processes struggle to account for the simultaneous appearance of all the necessary components. The intricate shape, active site, and catalytic properties of enzymes hint at intentional design rather than random chance. We must consider the limitations of mere chemical processes when unraveling the origins of such sophisticated biochemical systems. The probability of enzymes like adenylosuccinate synthase emerging through unguided events is staggeringly low. Given the intricate functional requirements and complex interactions involved, an intelligent designer offers a more plausible explanation for their origins. It is within the embrace of this perspective that we can appreciate the wonder and awe-inspiring complexity of adenylosuccinate synthase and other intricate biochemical systems. Adenylosuccinate synthase's choreography of phosphorylation, cleavage, activation, and condensation is a performance that orchestrates the synthesis of crucial nucleotides, while the ensemble of regulatory mechanisms ensures cellular harmony. And as we explore its origins, let us ponder the delicate balance between blind chemistry and the fingerprints of an intelligent designer.
Adenylosuccinate lyase (ASL)
In the intricate realm of cellular chemistry, Adenylosuccinate lyase (ASL) emerges as a remarkable performer. This enzyme takes on the task of cleaving adenylosuccinate—a crucial intermediate molecule—into AMP (adenosine monophosphate) and fumarate. This elegant reaction unfolds within the cytoplasm, marking the second step in the de novo synthesis of AMP. ASL's delicate choreography involves severing the bond between adenosine and succinate within adenylosuccinate. With this precise maneuver, AMP is born, poised to play its essential role in a myriad of cellular processes, from energy metabolism to RNA synthesis and signaling pathways. ASL, typically existing as a solitary entity, consists of a single polypeptide chain. Its structure embraces the captivating TIM barrel fold, a widely embraced motif in the world of enzymes. Named after the triosephosphate isomerase, the TIM barrel fold showcases an enchanting arrangement of alpha helices and beta strands, forming a barrel-like structure. This exquisite fold often boasts eight parallel strands of beta sheet interwoven with alpha helices, creating a rhythmic alternation around the core axis. The TIM barrel fold follows a choreographed pattern—strands adjacent, hands linked in loops or short helices. The grand finale of this structural symphony unfolds as the N- and C-termini of the protein unite, completing the barrel's poetic embrace. The interior of the TIM barrel, a sacred space, holds the enzyme's active site, rich in hydrophobic contours. This sanctum welcomes specific substrates and ligands, forming intimate interactions and enabling the enzyme's catalytic magic. The TIM barrel fold, with its versatile nature, captivates the scientific stage. Its diverse repertoire extends to enzymes performing vital roles in catalysis, isomerization, and metabolic pathways. This fold provides a stable and rigid framework, an unyielding canvas for the artist of catalysis to work their wonders. Its dance within the TIM barrel fold, a feat of structure and function, illuminates the intricate beauty of enzymes. In the grand theater of cellular chemistry, ASL takes center stage, painting a vivid picture of the wonders of life's molecular choreography.
Within the heart of Adenylosuccinate lyase (ASL), an active site brims with captivating activity. This enzyme possesses a unique talent—it acts specifically on adenylosuccinate, an exquisite substrate, cleaving it with precision to unveil the treasures within: AMP and fumarate. The active site of ASL serves as a stage for molecular interactions, where specific amino acid residues choreograph the enzyme's performance. Their strategic placement within the active site ensures the seamless execution of the enzymatic reaction, like a well-rehearsed dance. ASL's grand performance can be orchestrated by factors beyond its stage. At the transcriptional level, the expression of the gene responsible for producing this marvelous enzyme can be influenced by the cellular environment. The availability of purine nucleotides, those precious building blocks of life, can sway the expression of ASL and set the stage for its enzymatic prowess. Additionally, post-translational modifications and intricate interactions with other proteins can add nuances to ASL's performance, enhancing or modulating its activity. As a solitary performer, ASL thrives in its monomeric form. Its amino acid sequence, a blueprint etched in the language of life, ensures that the enzyme is poised for optimal function. Every letter in this sequence holds significance, contributing to the structural arrangement that allows ASL to unleash its catalytic powers with unrivaled efficiency. So let us marvel at the grand performance of Adenylosuccinate lyase, as it takes center stage, delicately binding to its adenylosuccinate partner and gracefully cleaving it to reveal the hidden treasures of AMP and fumarate. The expression of this captivating enzyme is finely tuned, responding to the needs of the cellular orchestra. In its monomeric glory, ASL dances through the intricacies of the enzymatic world, leaving us in awe of the harmonious interplay of molecules that underpins the symphony of life.
Last edited by Otangelo on Thu Jul 13, 2023 12:13 pm; edited 1 time in total