There are 3 classes of RNR enzymesThere are three classes of RNR enzymes, which differ in their structure and mechanism of action.
Class I RNR enzymes are found in eukaryotes, bacteria, and viruses. They use a radical mechanism to generate a stable tyrosyl radical on the enzyme, which then abstracts an electron from the substrate to initiate the reduction reaction. Class I RNR enzymes require a protein called R1 to provide the catalytic site for the reduction of ribonucleotides. R1 contains a dinuclear metal center composed of iron and tyrosine residues that are essential for the activity of the enzyme.
Class II RNR enzymes are found in bacteria and archaea. They use a different radical mechanism to generate a stable glycyl radical on the enzyme, which then abstracts an electron from the substrate. Unlike Class I RNR enzymes, Class II RNR enzymes do not require a separate protein for their activity, and the active site is located entirely within the enzyme.
Class III RNR enzymes are only found in aerobic bacteria and archaea. They use a radical mechanism similar to Class I RNR enzymes, but the reaction is initiated by a flavodoxin protein instead of a tyrosyl radical. Class III RNR enzymes are not well understood, and their function in these organisms is not yet clear.
E. Torrents (2014): Currently, three different RNR classes have been described (I, II, and III), and class I is further subdivided into Ia, Ib, and Ic. All three RNR classes share a common three-dimensional protein structure at the catalytic subunit and a highly conserved α/β barrel structure in the active site of the enzyme. In addition, the two potential allosteric centers (specificity and activity) are highly conserved among the different RNR classes, although in class Ib, and some class II RNRs activity allosteric site is absent.20
Daniel Lundin (2009):The significant differences between RNRs exist notably in cofactor requirements, subunit composition and allosteric regulation. These differences result in distinct operational constraints (anaerobicity, iron/oxygen dependence, and cobalamin dependence), and form the basis for the classification of RNRs into three classes. 22
T. B. Ruskoski (2021): More recently, remarkably diverse bioinorganic and radical cofactors have been discovered in class I RNRs from pathogenic microbes. These enzymes use alternative transition metal ions, such as manganese, or posttranslationally installed tyrosyl radicals for initiation of ribonucleotide reduction.21
Why are there 3 classes? While all three classes of RNR enzymes catalyze the same fundamental reaction of converting ribonucleotides to deoxyribonucleotides, they perform this reaction in different ways and under different conditions. This diversity is likely due to the varying environments and metabolic needs of the organisms in which these enzymes are found.
For example, Class I RNR enzymes are found in a wide range of organisms, including eukaryotes, bacteria, and viruses, and are essential for DNA replication and repair. Class II RNR enzymes, on the other hand, are only found in bacteria and archaea, and they are generally more resistant to oxidative stress than Class I enzymes, which may be important for their survival in harsh environments. Class III RNR enzymes are found only in aerobic bacteria and archaea and are thought to play a role in the regulation of iron homeostasis.
In addition, the different classes of RNR enzymes
have distinct structural and mechanistic features that may make them more or less suitable for different types of cellular processes. For example, Class I RNR enzymes require a separate protein for their activity, which may provide an additional level of regulation or allow for more precise control of the enzyme's activity. In contrast, Class II RNR enzymes are self-contained and may be better suited for rapid responses to changing environmental conditions.
Class II RNR enzymes found in bacteria and archaea function in environments with high levels of oxidative stress. Oxidative stress refers to an imbalance between the production of reactive oxygen species (ROS) and the cell's ability to detoxify them. ROS are highly reactive molecules that can damage cellular components such as proteins, lipids, and DNA, leading to cellular dysfunction and death. ROS are produced as byproducts of various metabolic processes, including respiration and photosynthesis, and their levels can increase in response to environmental stressors such as exposure to UV radiation or toxins.
Class II RNR enzymes found in bacteria and archaea are adapted to function in environments with high levels of oxidative stress. These enzymes have unique structural and mechanistic features that
allow them to withstand and repair the damage caused by ROS. For example, they have
a unique mechanism for generating a glycyl radical, which is used to initiate the reduction reaction and is highly resistant to oxidation. Additionally, Class II RNR enzymes have been shown to interact with antioxidant enzymes, such as thioredoxins and glutaredoxins, which can help to reduce ROS levels and prevent oxidative damage.
The ability of Class II RNR enzymes to function in environments with high levels of oxidative stress is
essential for the survival and adaptation of bacteria and archaea in harsh environments. Reducing ROS is an additional function of Class II RNR enzymes, in addition to their primary function of transforming RNA to DNA. They can be considered
multifunctional enzymes, as they have more than one function in the cell. Some studies have suggested that Class II RNR enzymes may have a role in the regulation of gene expression, particularly in response to stress or nutrient availability. Other studies have suggested that Class II RNR enzymes may have a role in the production of secondary metabolites or in the metabolism of xenobiotics (foreign compounds). However, these proposed functions are still the subject of ongoing research and are not yet fully understood.
What are the environments that require organisms with Class II RNR enzymes? Class II RNR enzymes are required by organisms that live in harsh environments where oxygen is scarce or absent, such as
deep-sea hydrothermal vents, anaerobic sediments, or inside the guts of some animals. These environments are characterized by low levels of oxygen, high levels of toxic compounds, extreme temperatures, and high pressures.
In these environments, Class II RNR enzymes are necessary for the synthesis of deoxyribonucleotides, the building blocks of DNA. These enzymes use a different mechanism than Class I RNR enzymes to generate the free radical needed to initiate the reaction, which does not require oxygen. This allows organisms to synthesize DNA even in the absence of oxygen.
Some examples of organisms that require Class II RNR enzymes include anaerobic bacteria such as Clostridium species, which live in the gut of animals and are involved in the breakdown of organic matter, and archaea such as Pyrococcus furiosus, which live in hot environments such as deep-sea hydrothermal vents and use Class II RNR enzymes to synthesize DNA in the absence of oxygen.
How are Class II RNR enzymes distinct from the other two classes?Class II RNR enzymes are distinct from the other two classes (Class I and Class III) in several ways:
Structure: Class II RNR enzymes have a completely different protein structure than Class I and III RNR enzymes. They consist of a single protein subunit, unlike Class I and III RNR enzymes, which are composed of multiple subunits.
Oxygen-independent: Class II RNR enzymes do not require oxygen to generate the free radical needed to initiate the reaction, unlike Class I RNR enzymes which use oxygen as a co-substrate, and Class III RNR enzymes which require a protein called AdoCbl (Adenosylcobalamin) to generate the free radical.
Metallocofactor: Class II RNR enzymes contain a different metallocofactor (metal ion-containing non-protein component) than the other two classes. Specifically, Class II RNR enzymes use a non-heme iron center with a tyrosyl radical, whereas Class I RNR enzymes use a di-iron center, and Class III RNR enzymes use AdoCbl.
Class II RNR enzymes are believed to have a different origin than the other two classes. Class II RNR enzymes
appear to have a distinct origin and are only found in certain bacterial and archaeal species.
These enzymes are highly resistant to oxidation and
have unique features that allow them to function in such conditions. In contrast, Class I RNR enzymes found in eukaryotes, bacteria, and viruses are optimized to function in environments with lower levels of oxidative stress and have different mechanisms to facilitate their activity. Similarly, Class III RNR enzymes are specialized to function in aerobic bacteria and archaea and have
unique mechanisms to allow them to operate in these environments.
The differences in the environments in which these enzymes function can be attributed to a variety of factors, including the presence of different reactive oxygen species, variations in pH and temperature, and variations in the availability of cofactors and substrates. The mechanisms employed by each class of RNR enzymes have evolved to optimize their activity in their respective environments and to ensure that they can perform their essential functions under the appropriate conditions.
In summary, the unique features of the different classes of RNR enzymes allow them to function optimally in different environments, depending on the organism in which they are found. This specialization is necessary for the efficient and effective operation of cellular processes and highlights the importance of the environment in shaping the function and evolution of biological molecules.
Independent origin of the three RNR classesClass II RNR enzymes most likely do not share a common ancestor with the other two classes of RNR enzymes, but emerged separately. This is supported by several lines of evidence, including their distinct protein structure, metallocofactor, and mechanism of action. 28 The history and origin of RNR enzymes are intimately connected to the broader question of the origin of life on Earth since these enzymes play a critical role in the synthesis and maintenance of genetic material in all living organisms, The three classes of Ribonucleotide Reductase (RNR) enzymes most likely
have an independent and unique trajectory of origin. Class I RNR enzymes use a metallocofactor, a diferric-tyrosyl radical, to catalyze the conversion of nucleotides into deoxynucleotides. Class II RNR enzymes have a different structure and mechanism than the other two classes. They use a stable tyrosyl radical, rather than a diferric-tyrosyl radical, to initiate the nucleotide reduction reaction. Class III RNR enzymes were discovered relatively recently and have a unique mechanism that involves a stable glycyl radical and do not require any metals or cofactors for activity. The mechanisms of horizontal gene transfer, gene duplication, and convergent evolution are not adequate to explain all of the dissimilarities between the three classes of RNR enzymes.
The three metal RNR Co-factorsClass I of Ribonucleotide reductases occurs in aerobically thriving organisms including humans uses oxygen activated by a dinuclear iron center to convert a tyrosine residue into a radical. Class II is the coenzyme B12-dependent reductaseClass III contains an extremely oxygen-sensitive glycyl radical, which is generated with the aid of S-adenosylmethionine (SAM). All three types, however, use a thiyl radical at the active site and act by an almost identical mechanism.
All three types use a thiyl radical at the active site and act by an almost identical mechanism.Lander, E. S (2001): Different classes of RNR's have intriguing sequence “motifs” involving cysteines that appear to be important for the catalysis (in Escherichia coli, Cys-439, the radical site, and Cys-225 and Cys-462, which delivers two electrons and a proton). These motifs offer tantalizing suggestions that all RNRs are related by common ancestry but underwent divergent evolution so massive that only traces of evidence for homology remain in the sequences themselves. These motifs are inadequate to provide a statistically significant case for homology, however, and motifs are notoriously inadequate for confirming homology in general. 29Comment: To claim common ancestry, in this case, is an ad-hoc assertion. Truth said, science is unable to infer a reasonable scenario out of the evidence, and all it can do, is resort to made-up stories, which bear no credibility. The best and most straightforward explanation is that a creator made RNRs, and equipped each of them with different ways to perform the same function.
RNR structureClass I RNR enzymes Class I ribonucleotide reductase (RNR) enzymes are composed of two subunits, RRM1 and RRM2.
RRM1 subunit is a large protein consisting of approximately 800 amino acids. It contains two domains, the N-terminal domain and the C-terminal domain. The N-terminal domain is responsible for binding to the small subunit (RRM2) and contains a zinc finger motif that is involved in protein-protein interactions. The C-terminal domain contains the active site for ribonucleotide reduction and is composed of two subdomains: the substrate-binding subdomain and the radical-generating subdomain.
RRM2 subunit is a smaller protein consisting of approximately 350 amino acids. It contains a single domain with a unique structure known as the RNR-specificity loop. This loop is responsible for determining the specificity of the enzyme for the different ribonucleotides and contains a conserved tyrosine residue that plays a critical role in the catalytic mechanism of the enzyme.
The tertiary structure of the RNR enzyme is complex and involves the interaction of the two subunits. RRM1 and RRM2 form a heterodimeric enzyme complex that is regulated by the binding of different allosteric effectors. In the absence of allosteric effectors, RNR is in an inactive state, but upon binding of allosteric effectors, the enzyme complex undergoes conformational changes that allow for activation of the enzyme and subsequent ribonucleotide reduction.
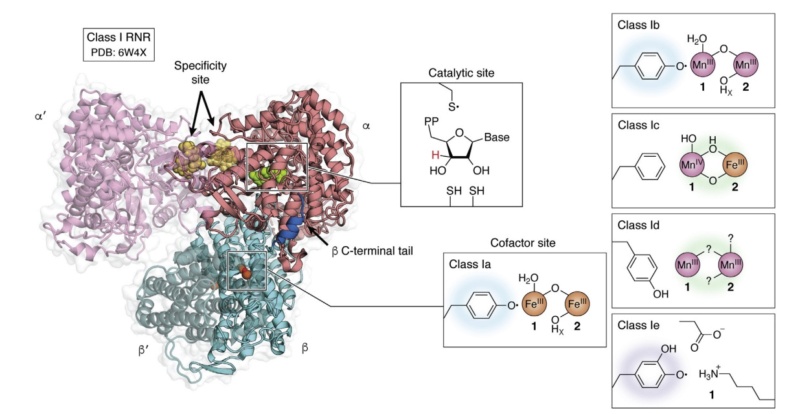 Quaternary structure of the active holoenzyme complex in class I RNR
Quaternary structure of the active holoenzyme complex in class I RNR (PDB accession code 6W4X). Insets show the location of the active site in the catalytic α subunit (middle top) and the metallo- or radical cofactor (middle bottom and far right) in the β subunit. 30
Overall, the structure of class I RNR enzymes is highly conserved across species and is essential for the de novo synthesis of deoxyribonucleotides, which are critical for DNA synthesis and cell division.
Class II RNR enzymes The structure of class II RNR enzymes is unique and distinct from class I enzymes. These enzymes are composed of a large α subunit and a smaller β subunit, and the two subunits form a stable heterodimeric complex.
The α subunit of class II RNR enzymes contains two domains: the N-terminal domain and the C-terminal domain. The N-terminal domain contains the di-iron center, which is responsible for ribonucleotide reduction. The C-terminal domain is responsible for binding to the β subunit and contains a loop structure known as the specificity loop, which determines the specificity of the enzyme for different ribonucleotides.
The β subunit of class II RNR enzymes contains a single domain that is responsible for binding to the α subunit. It contains a conserved cysteine residue that is involved in the regulation of the enzyme.
The tertiary structure of class II RNR enzymes is highly conserved across species and is critical for enzyme function. The α and β subunits form a heterodimeric enzyme complex that is regulated by several allosteric effectors. Binding of allosteric effectors causes conformational changes in the enzyme complex that allow for activation of the enzyme and subsequent ribonucleotide reduction.
Overall, the structure of class II RNR enzymes is unique and distinct from class I enzymes, but both classes of enzymes are essential for the de novo synthesis of deoxyribonucleotides and DNA replication.
Class III RNR enzymes Class III ribonucleotide reductase (RNR) enzymes are found in bacteriophages and some bacteria. These enzymes use a glycyl radical to reduce ribonucleotides, similar to class I RNR enzymes.
The structure of class III RNR enzymes is unique and distinct from class I and class II enzymes. These enzymes are composed of a single polypeptide chain that contains three domains: the N-terminal domain, the central domain, and the C-terminal domain.
The N-terminal domain of class III RNR enzymes contains a glycyl radical that is essential for enzyme function. The glycyl radical is generated by a radical SAM (S-adenosylmethionine) enzyme and is stabilized by the protein environment.
The central domain of class III RNR enzymes contains a conserved cysteine residue that is involved in the regulation of the enzyme.
The C-terminal domain of class III RNR enzymes contains a cluster of iron-sulfur (Fe-S) clusters that are involved in electron transfer and ribonucleotide reduction.
The tertiary structure of class III RNR enzymes is critical for enzyme function and involves the interaction of the three domains. The glycyl radical in the N-terminal domain is stabilized by the protein environment, and the central and C-terminal domains are involved in electron transfer and ribonucleotide reduction.
Overall, the structure of class III RNR enzymes is unique and distinct from class I and class II enzymes, but all three classes of enzymes are essential for the de novo synthesis of deoxyribonucleotides and DNA replication.
While all three classes of RNR enzymes share a common function, their proteic architecture is quite distinct from one another.
RNR uses radical chemistry to catalyze the reduction of each NTP. How the enzyme generates this radical, the type of cofactor and metal required, the three-dimensional structure of this enzyme complex and the dependence of oxygen are all characteristics that are considered when classifying RNRs.The X-ray structures of the R1 catalytic component and the R2 di-iron component of the class I RNR from E. coli were determined . The catalytic subunit was found to be a novel 10-stranded α/β-barrel with a loop that hosts the thiyl
radical protruding into its center. Despite a lack of sequence similarity with other ribonucleotide reductases, all ribonucleotide reductases would have similar catalytic subunits, reflecting their similar catalytic strategies. The structure of the catalytic subunit of the class III enzyme revealed the characteristic 10-stranded α/β-barrel with a central loop bearing the thiyl radical precursor. Many features of the structure reported are remarkably similar to the structures of the catalytic subunits of the class I and class III enzymes, even though there is <10% sequence homology among them. The similarities and contrasts with the enzymes of other classes have much to tell us about all ribonucleotide reductases.Comment: The three classes of RNR enzymes have the same catalytic activity, but different amino acid sequences to reach the same result. Science has no good explanations for the divergence.
RNR Mechanism and reactionThe mechanism of ribonucleotide reductase (RNR) can vary depending on the class of the enzyme. Each of the three classes has a distinct mechanism for converting ribonucleotides to deoxyribonucleotides.
The mechanism in Class I RNR enzymes Class I RNR enzymes use a free radical mechanism. The free radical in RNR enzymes is generated through a specific reaction that involves the reduction of a disulfide bond in the enzyme's active site by a cysteine residue. The reaction in which the free radical in RNR enzymes is generated
is a multi-step process that can be divided into two stages: initiation and propagation.
Initiation:The RNR enzyme contains a disulfide bond between two cysteine residues in the active site. The two cysteine residues in the active site of RNR enzymes are typically recruited from the enzyme's own polypeptide chain. During the synthesis of the enzyme, the amino acid sequence of the polypeptide chain includes these cysteine residues in a specific location within the enzyme's three-dimensional structure, which ultimately forms the enzyme's active site. In some cases, the cysteine residues may be supplied by a separate protein that interacts with the RNR enzyme, but this is less common.
A reducing agent (such as thioredoxin) transfers an electron to the disulfide bond, causing it to break and generating two separate cysteine residues, each with a single unpaired electron (also known as thiyl radicals).
One of the thiyl radicals is rapidly converted into a stable thiol group by reaction with a nearby protein cysteine residue, which helps to prevent unwanted reactions.
Propagation:One of the thiyl radicals (Cys•) on the enzyme reacts with molecular oxygen to generate a peroxide intermediate (Cys-S-O-O•).
The peroxide intermediate is then rapidly converted into a tyrosyl radical (Tyr•) on a nearby tyrosine residue by an electron transfer reaction.
The tyrosyl radical is then transferred to a substrate molecule (such as a ribonucleotide diphosphate), which initiates the radical-mediated chemistry necessary for nucleotide reduction.
Overall, the generation of the free radical in RNR enzymes
is a carefully orchestrated process that allows for precise control over the production of reactive species, enabling the enzyme to carry out its essential functions in DNA synthesis and repair.
Once the two cysteine amino acids in the active site of the RNR enzyme have been used in the reaction to generate the free radical, they are converted to a disulfide bond. This disulfide bond then needs to be reduced in order for the enzyme to continue functioning. In class I RNR enzymes, this reduction is accomplished by a flavoprotein known as thioredoxin reductase, which transfers electrons from NADPH to a molecule of thioredoxin. Thioredoxin then reduces the disulfide bond in the RNR enzyme's active site, regenerating the cysteine residues and allowing the enzyme to continue its catalytic cycle. In class II and III RNR enzymes, different electron transfer proteins are involved in the reduction of the disulfide bond.
This reduction leads to the formation of a thiyl radical on the cysteine residue and a transient tyrosyl radical on a nearby tyrosine residue. A thiyl radical is a highly reactive species that contains an unpaired electron on the sulfur atom of a cysteine residue. It is formed in the RNR enzyme during the process of generating the free radical required for the enzyme's catalytic activity. The thiyl radical plays a critical role in the enzyme's mechanism by abstracting a hydrogen atom from the substrate, thereby initiating the radical transfer process that leads to the generation of deoxyribonucleotides. The tyrosyl radical is then transferred to a substrate molecule, which initiates the radical-mediated chemistry necessary for nucleotide reduction. This radical transfer process is what makes RNR enzymes unique and essential for DNA synthesis and repair in all living organisms.
The active site of the enzyme contains a tyrosine residue that is used to generate a free radical on the ribonucleotide. The process occurs in several steps:
1. A substrate (the ribonucleotide) binds to the enzyme's active site, where it is coordinated by several amino acid residues, including the tyrosine residue.
2. An adjacent cysteine residue in the active site donates an electron to the tyrosine residue, creating a tyrosyl radical.
3. The tyrosyl radical abstracts a hydrogen atom from the substrate, creating a substrate radical and regenerating the tyrosine residue.
4. The substrate radical then reacts with the thiyl radical generated from the cysteine residue in the earlier step, resulting in the formation of a new covalent bond between the ribonucleotide and the cysteine residue.
5. This reaction produces a new cysteine residue with a thiol group, and a new substrate that has been converted to its corresponding deoxyribonucleotide.
6. Overall, the tyrosine residue acts as a mediator, transferring the radical to the substrate to enable the reduction reaction to occur.
The essential players involved in the process to generate a free radicalGenerating a free radical on the ribonucleotide in RNR enzymes requires:
1. The RNR enzyme itself contains the active site responsible for the generation of the free radical.
2. A source of electrons, which reduces the disulfide bond in the enzyme's active site. In class I and II RNRs, this source is a flavoprotein that donates electrons to the enzyme. In class III RNRs, the source is a ferredoxin.
3. The substrate ribonucleotide, which is targeted by the free radical and converted into its corresponding deoxyribonucleotide form.
4. The amino acid residues in the enzyme's active site, including the cysteine and tyrosine residues, which are essential for the formation and stabilization of the free radical.
All of these components are necessary for the RNR enzyme to function properly and carry out its crucial role in DNA synthesis and repair. The RNR enzyme can be considered irreducibly complex, as it requires the coordinated and functional interaction of multiple components to generate the free radical necessary for DNA synthesis and repair. Removal or impairment of any one of these components would render the enzyme non-functional. The individual players/subunits/substrates involved in the process of generating a free radical on the ribonucleotide in RNR enzymes would have no function on their own, and they need to be integrated in the system for the enzyme to function properly. This is a key characteristic of irreducible complexity, where the individual components of a complex system are interdependent and cannot function on their own. The probability of the individual players arising through purely random, unguided processes is warranted to be considered low to the extreme, even by many scientists due to the complexity and specificity of these systems.
Dr. Douglas Axe, a molecular biologist and director of the Biologic Institute, has written extensively on the subject of protein evolution:
"The kind of enzyme we're talking about here is mind-bogglingly complex. It's a gigantic machine. It's not just a couple of amino acids strung together. You're talking about a machine that has multiple moving parts, has different metals, it has different ligands that it has to bind to. It has to be regulated. It's an incredibly complex thing." ( he was not referring to the ribonucleotide reductase specifically, but he was speaking more generally about the complexity of certain enzymes, including many proteins involved in cellular metabolism. )
Dr. Axe made this statement in a 2016 interview with The College Fix, in which he discussed his research on protein evolution and his skepticism of the idea that complex proteins like RNR enzymes could have arisen by chance through naturalistic processes.
The mechanism in Class II RNR enzymes The Class II RNR enzyme is a homodimer, meaning it consists of two identical subunits. Each subunit contains three domains: a substrate-binding domain, a radical-generating domain, and a catalytic domain.
1. The substrate-binding domain of each subunit binds to a ribonucleotide, specifically the 2'-OH group of the ribose sugar.
2. The radical-generating domain of each subunit contains a cofactor called adenosylcobalamin (AdoCbl), which is a form of vitamin B12. The AdoCbl is converted to a highly reactive species called 5'-deoxyadenosyl radical (dAdo•) by the transfer of an electron from a nearby iron-sulfur cluster.
3. The dAdo• radical is then transferred from one subunit to the other, across the dimer interface, where it reacts with the ribonucleotide bound to the substrate-binding domain. The dAdo• radical abstracts a hydrogen atom from the 2'-OH group of the ribose sugar, generating a carbon-centered radical on the sugar ring.
4. The carbon-centered radical is then stabilized by the radical-generating domain, which donates an electron to the radical, converting it to a stable intermediate.
5. The stable intermediate is then transferred to the catalytic domain, where it undergoes a series of proton and electron transfers, leading to the reduction of the ribonucleotide to a deoxyribonucleotide.
6. Finally, the deoxyribonucleotide product is released, and the enzyme returns to its starting state, ready to bind to another ribonucleotide substrate and repeat the cycle.
The mechanism of Class II RNR enzymes is highly complex and involves multiple subunits, cofactors, and radical intermediates.
Class II RNR enzymes differ from the other two classes of RNR enzymes (Class I and Class III) in both their structure and mechanism. The most notable structural difference is that Class II RNR enzymes are homodimers, meaning that they consist of two identical subunits, while Class I and III RNR enzymes are heterodimers, meaning they consist of two different subunits. In terms of mechanism, Class II RNR enzymes use a radical-based mechanism, while Class I and III RNR enzymes use a different mechanism that involves the formation of a free radical on a cysteine residue in the active site of the enzyme. In Class II RNR enzymes, the radical is generated on a cofactor called adenosylcobalamin (AdoCbl), while in Class I and III RNR enzymes, the radical is generated on a conserved cysteine residue. Another difference between the three classes of RNR enzymes is the way in which they are regulated. Class II RNR enzymes are typically regulated at the level of gene expression, meaning that their activity is controlled by the production or degradation of the enzyme itself. In contrast, Class I and III RNR enzymes are regulated by a variety of mechanisms, including allosteric regulation, protein-protein interactions, and post-translational modifications. While all three classes of RNR enzymes catalyze the conversion of ribonucleotides to deoxyribonucleotides, they differ in their structural features, reaction mechanisms, and modes of regulation.
The complexity of Class II RNR enzymes presents a challenge to understanding how they could have evolved from simpler precursors. One suggestion of evolutionary relatedness to the other versions is the fact that both, Class I and Class II RNR enzymes contain an iron-sulfur cluster, and both use a radical-generating cofactor to initiate nucleotide reduction. The iron-sulfur clusters in Class I and Class II RNR enzymes are similar, but not identical. Both classes of enzymes use iron-sulfur clusters to transport electrons during the nucleotide reduction process, but the specific structures and functions of these clusters differ between the two classes.
Comparing the iron-sulfur cluster between Class I, and Class II RNR enzymesIron-sulfur clusters are found in a wide range of proteins in almost all forms of life, including bacteria, archaea, and eukaryotes. There are some cells or organisms that do not contain enzymes or proteins with iron-sulfur clusters, either because they do not require them for their metabolic processes or because they have evolved alternative mechanisms for performing the same functions. For example, some anaerobic bacteria can use other types of electron carriers, such as flavoproteins or quinones, instead of iron-sulfur clusters for their energy metabolism. In addition, some organisms may have evolved different mechanisms for DNA repair and other cellular processes that do not rely on iron-sulfur clusters. Iron-sulfur clusters are highly versatile and are involved in a wide range of cellular processes, and they are considered to be one of the oldest and most conserved cofactors in biology. Therefore, it is unlikely that cells or organisms could completely do without iron-sulfur clusters or an equivalent mechanism to carry out their essential metabolic processes. The origin of iron-sulfur clusters is considered to be an origin of life problem. Iron-sulfur clusters are one of the oldest and most widespread cofactors in biology, and they are found in a wide range of proteins involved in various cellular processes, including energy metabolism, DNA replication and repair, and regulation of gene expression.
The "iron-sulfur world" hypothesisThe "iron-sulfur world" hypothesis is a theory regarding the origin of life on Earth that suggests that life may have originated in an environment rich in iron and sulfur minerals. This hypothesis proposes that the first living organisms may have used iron-sulfur clusters as a primitive form of enzymatic activity, which could have facilitated the chemical reactions necessary for the emergence of life.
The iron-sulfur world hypothesis is based on several observations. First, iron and sulfur are abundant elements that were likely present in the early Earth's crust and oceans. Second, iron-sulfur clusters are highly versatile and can catalyze a wide range of chemical reactions, including those involved in energy metabolism, DNA replication and repair, and the synthesis of amino acids and other organic molecules. Third, iron-sulfur clusters are highly conserved in modern organisms, suggesting that they may have been present in the last universal common ancestor (LUCA) of all life forms.
According to the iron-sulfur world hypothesis, the first living organisms would have used iron-sulfur clusters to carry out primitive forms of metabolic and enzymatic activity, which could have allowed them to harness the energy and resources available in the early Earth's environment. Over time, these organisms would have generated more complex metabolic pathways and biochemical processes, leading to the emergence of the diverse forms of life that exist today.
Iron-sulfur clusters Class I RNR enzymesIn Class I RNR enzymes, the iron-sulfur cluster is a [Fe-S] cluster that consists of two iron ions and two sulfur atoms coordinated by cysteine residues in the protein. This cluster serves as an electron carrier, transferring electrons from the radical-generating cofactor to the active site of the enzyme where nucleotide reduction occurs.
In contrast, the iron-sulfur cluster in Class II RNR enzymes is a [Fe4S4] cluster that consists of four iron ions and four sulfur atoms coordinated by cysteine residues. This cluster is also involved in electron transport during nucleotide reduction, but its structure and function differ from that of the [Fe-S] cluster in Class I RNR enzymes.
Furthermore, the biosynthesis pathways for the iron-sulfur clusters in Class I and Class II RNR enzymes are similar in some respects, but differ in others.
In Class I RNR enzymes, the [Fe-S] cluster is synthesized by a complex set of enzymes called the NifS/NifU system. This system involves the transfer of sulfur from cysteine to a scaffold protein, followed by the insertion of iron ions to form the complete cluster. The [Fe-S] cluster is then incorporated into the RNR enzyme during its maturation process.
Iron-sulfur clusters Class I RNR enzymesIn Class II RNR enzymes, the [Fe4S4] cluster is also synthesized by the NifS/NifU system, but the assembly process is more complex. In addition to the transfer of sulfur from cysteine to the scaffold protein, the assembly of the [Fe4S4] cluster requires the involvement of several accessory proteins. These proteins are thought to help with the coordination of the iron ions and the formation of the cluster structure. Once the [Fe4S4] cluster is assembled, it is incorporated into the RNR enzyme during maturation.
Overall, the biosynthesis pathways for the iron-sulfur clusters in Class I and Class II RNR enzymes are complex and involve multiple steps and protein components. While there are similarities between the two pathways, the differences in the structures of the two clusters mean that there are also significant differences in the details of their biosynthesis.
The biosynthesis of the iron-sulfur cluster in Class I RNR enzymes involves
multiple enzymes and protein components. The exact number of enzymes involved can vary depending on the organism and the specific details of the biosynthetic pathway, but typically there are at least three enzymes involved in the process. The first enzyme is called NifS, which is responsible for transferring sulfur from cysteine to a specialized scaffold protein called IscU. IscU then binds iron ions and facilitates their incorporation into the growing iron-sulfur cluster. The second enzyme involved in the process is NifU, which serves as a scaffold for the assembly of the iron-sulfur cluster. NifU interacts with IscU and other proteins to coordinate the incorporation of sulfur and iron ions into the cluster. Finally, the third enzyme involved in the process is a specialized chaperone protein called HscA/HscB. This protein helps to prevent the premature aggregation of the nascent iron-sulfur cluster and ensures its proper folding and incorporation into the RNR enzyme. Other proteins may also be involved in the biosynthesis of the Class I RNR iron-sulfur cluster, and the specific details of the process can vary depending on the organism and environmental conditions. The simplest biosynthesis pathway for the Class I RNR iron-sulfur cluster involves two enzymes: NifS and IscA. NifS transfers sulfur to IscA, which then binds iron ions to form the [Fe-S] cluster. The [Fe-S] cluster is then incorporated into the RNR enzyme during its maturation process.
The biosynthesis of the iron-sulfur cluster in Class II RNR enzymes involves more players than in Class I RNR enzymes. The exact number of players involved can vary depending on the organism and the specific details of the biosynthetic pathway, but typically there are at least six proteins involved in the process.
The first enzyme involved in the process is NifS, which transfers sulfur to a protein called IscA.
The second enzyme is called SufB, which interacts with IscA to assemble a [2Fe-2S] cluster.
The third enzyme is called SufC, which binds the [2Fe-2S] cluster and then interacts with SufB to assemble a [4Fe-4S] cluster.
The fourth enzyme is called SufD, which binds the [4Fe-4S] cluster and then interacts with SufC to facilitate its transfer to the RNR enzyme.
The fifth protein involved in the process is called SufA, which helps to transfer the [4Fe-4S] cluster from SufD to the RNR enzyme.
Finally, a sixth protein called SufE has also been implicated in the process, although its exact role is not yet fully understood.
The biosynthesis pathway for the iron-sulfur cluster in Class II RNR enzymes is complex and involves multiple steps and protein components. The involvement of multiple proteins in the process likely reflects the greater complexity of the [4Fe-4S] cluster itself.
Quality control in producing the iron-sulfur clustersRNR enzymes contain Fe-S clusters, and errors in Fe-S cluster synthesis or assembly can lead to enzyme dysfunction and impaired DNA synthesis. Therefore, the error check process for Fe-S cluster synthesis in RNR enzymes is critical for maintaining proper enzyme function.
One of the ways that cells prevent errors in Fe-S cluster synthesis in RNR enzymes is through a protein called NrdH-redoxin, which serves as a chaperone for the Fe-S clusters during their assembly. NrdH-redoxin helps to prevent misincorporation of iron or sulfur atoms into the Fe-S cluster, which could result in impaired enzyme function.
Another mechanism for error-checking Fe-S cluster synthesis in RNR enzymes is through the use of iron-responsive element (IRE) sequences in the messenger RNA (mRNA) that encodes the RNR enzyme. IRE sequences are recognized by iron regulatory proteins (IRPs), which can bind to the mRNA and regulate its translation into protein. If the cell detects an error in Fe-S cluster synthesis, IRPs can block translation of the mRNA, preventing the synthesis of defective RNR enzymes.
Finally, cells can also use quality control mechanisms to monitor the activity of RNR enzymes with Fe-S clusters. For example, cells may increase the expression of other Fe-S cluster-containing proteins to compensate for impaired RNR enzyme function, or they may activate stress response pathways to help the cell cope with the effects of defective Fe-S clusters.
Another mechanism for error-checking Fe-S cluster synthesis in RNR enzymes is through the use of iron-responsive element (IRE) sequences in the messenger RNA (mRNA) that encodes the RNR enzyme. IRE sequences are recognized by iron regulatory proteins (IRPs), which can bind to the mRNA and regulate its translation into protein. If the cell detects an error in Fe-S cluster synthesis, IRPs can block translation of the mRNA, preventing the synthesis of defective RNR enzymes.
Cells can also use quality control mechanisms to monitor the activity of RNR enzymes with Fe-S clusters. For example, cells may increase the expression of other Fe-S cluster-containing proteins to compensate for impaired RNR enzyme function, or they may activate stress response pathways to help the cell cope with the effects of defective Fe-S clusters.
In both Class I and Class II RNR enzymes, the quality control mechanism involves as well a protein called SufBCD, which
recognizes and degrades iron-sulfur clusters that are improperly assembled or damaged. SufBCD acts as a "proofreading" mechanism, checking the quality of the iron-sulfur cluster before it is incorporated into the RNR enzyme. If the cluster fails the quality control check, it is disassembled and its components are recycled to prevent their incorporation into the RNR enzyme.
Overall, the quality control mechanism in the biosynthesis of iron-sulfur clusters in RNR enzymes is an important safeguard that helps to ensure the proper function of these enzymes in maintaining genome integrity and preventing DNA damage.
The proper synthesis and function of RNR enzymes, as well as many other biological molecules and systems, requires in most, if not in all cases, the implementation of error check and repair systems from the start. Without these systems, the error rate would likely be too high for the enzyme or system to function properly, potentially leading to cellular damage, disease, or even death. The existence of error check and repair systems are often interdependent with other biological processes, such as the synthesis and function of RNR enzymes, DNA replication and repair, and many other processes. In many cases, the proper functioning of these biological processes relies on the presence of error check and repair systems to maintain their integrity and prevent damage or errors from occurring. This interdependence between different biological processes is a common feature of living organisms and is often powerful evidence for the necessity of a mind with foreknowledge and foresight to instantiate such complexity and sophistication in biological systems.
SufBCD, the error check and repair machine in the cellSufBCD is a protein complex involved in the biogenesis of iron-sulfur clusters, which are important cofactors found in a wide range of proteins involved in various cellular processes, including electron transport, DNA replication and repair, and regulation of gene expression. The SufBCD complex is composed of three subunits: SufB, SufC, and SufD. SufB is a peripheral membrane protein that interacts with the inner membrane of bacteria, while SufC and SufD are cytoplasmic proteins. SufB contains a conserved domain that is involved in binding iron-sulfur clusters, while SufC and SufD interact with each other to form a nucleotide-binding domain that binds ATP and helps to regulate the activity of the complex. The complex also interacts with other proteins involved in iron-sulfur cluster biogenesis, such as SufA and SufE, to
facilitate the transfer and incorporation of the iron-sulfur clusters into target proteins.The SufBCD complex is important for the survival of bacteria in environments with limited iron availability, as iron-sulfur clusters are necessary for the activity of many essential enzymes involved in metabolism and other cellular processes. Dysfunction or deficiency of the SufBCD complex can lead to impaired iron-sulfur cluster biogenesis and various cellular defects, including sensitivity to oxidative stress, DNA damage, and antibiotic treatments. The SufBCD complex is composed of three subunits: SufB, SufC, and SufD. SufB is a peripheral membrane protein that interacts with the inner membrane of bacteria, while SufC and SufD are cytoplasmic proteins.
SufB contains a conserved domain that is involved in binding iron-sulfur clusters, while SufC and SufD interact with each other to form a nucleotide-binding domain that binds ATP and helps to regulate the activity of the complex.
The action core of the SufBCD complex is the SufB subunit, which contains a conserved domain that is involved in the binding of iron-sulfur clusters. This domain is known as the Fe-S cluster-binding domain, and it contains three cysteine residues that are involved in coordinating the binding of the iron and sulfur ions that make up the cluster. The Fe-S cluster-binding domain is located near the N-terminus of the SufB protein and is essential for the function of the complex. The co-factor of the SufBCD complex is the iron-sulfur cluster, which is a small, inorganic molecule composed of iron and sulfur ions. Iron-sulfur clusters are essential cofactors found in many proteins involved in cellular processes, including energy metabolism, DNA replication and repair, and regulation of gene expression. They are known for their ability to transfer electrons and to act as redox centers in many enzymatic reactions.
The biosynthesis of iron-sulfur clusters occurs through a complex pathway that involves several proteins, including the SufBCD complex. In this pathway, sulfur and iron ions are imported into the cell through various transport systems and are assembled into iron-sulfur clusters by the action of specific proteins. The SufBCD complex is involved in the later stages of this pathway, where it helps to transfer the iron-sulfur clusters to target proteins, where they can be incorporated into the active sites of enzymes. The SufBCD complex is an essential component of the iron-sulfur cluster biosynthesis pathway, and plays a critical role in the assembly and transfer of iron-sulfur clusters to target proteins.
The synthesis pathway to make SufBCDThe SufBCD enzyme is composed of three different subunits: SufB, SufC, and SufD. The biosynthesis pathway of the SufBCD enzyme involves the
coordinated expression and assembly of these subunits, as well as the synthesis and incorporation of the iron-sulfur clusters that are required for its function. Here is an overview of the synthesis pathway of the SufBCD enzyme:
1. Transcription: The genes encoding the SufBCD subunits are transcribed from the DNA into messenger RNA (mRNA) by the RNA polymerase enzyme.
2. Translation: The mRNA is then translated into protein by ribosomes, with each subunit being synthesized separately.
3. Chaperone proteins: As the individual subunits are synthesized, they are bound and stabilized by chaperone proteins, which prevent them from aggregating and ensure proper folding.
4. Assembly: Once all three subunits have been synthesized and properly folded, they are assembled into the SufBCD enzyme complex. This assembly process is coordinated by a series of accessory proteins, which help to ensure that the subunits are correctly positioned and oriented relative to each other.
Iron-sulfur cluster synthesis: The final step in the biosynthesis of the SufBCD enzyme involves the synthesis and incorporation of the iron-sulfur clusters that are required for its function. This process is mediated by a separate set of accessory proteins, which help to guide the assembly of the clusters and ensure that they are properly incorporated into the subunits of the enzyme. Overall, the biosynthesis pathway of the SufBCD enzyme is a complex and tightly regulated process that involves the coordinated expression, folding, and assembly of multiple protein subunits, as well as the synthesis and incorporation of the iron-sulfur clusters that are required for its function.
The accessory proteins involved in the synthesis of the Iron-sulfur cluster used in SufBCDThe biosynthesis of iron-sulfur clusters and their incorporation into proteins like SufBCD is a complex process that involves the coordination of multiple accessory proteins. Here is an overview of some of the accessory proteins involved in this process:
1. SufA: This protein is an iron-sulfur cluster scaffold protein that helps to assemble the iron-sulfur clusters that are required for the function of the SufBCD enzyme. SufA binds to the iron and sulfur atoms and helps to coordinate their assembly into a stable cluster.
2. SufE: This protein is an iron-sulfur cluster carrier protein that helps to deliver the clusters from the SufA scaffold protein to the target proteins like SufBCD. SufE binds to the clusters and then interacts with other proteins to facilitate their transfer.
3. SufB: This is one of the subunits of the SufBCD enzyme itself, but it also functions as an accessory protein during the biosynthesis of iron-sulfur clusters. SufB helps to deliver iron and sulfur to the SufA scaffold protein, and also interacts with SufC and SufD to coordinate the assembly of the iron-sulfur clusters within the SufBCD complex.
4. SufC: This is another subunit of the SufBCD enzyme, and it plays a critical role in the biosynthesis of iron-sulfur clusters by interacting with both SufB and SufD to coordinate the assembly of the clusters within the enzyme complex.
Overall, the biosynthesis of iron-sulfur clusters and their incorporation into proteins like SufBCD is a complex process that requires the coordination of multiple accessory proteins. These accessory proteins help to ensure that the clusters are assembled and delivered to their target proteins in a precise and controlled manner, which is essential for the proper function of the enzyme.
Another chicken & egg problemThere is another chicken-and-egg problem when it comes to the biosynthesis of iron-sulfur clusters since many enzymes that are involved in the synthesis and incorporation of these clusters themselves require iron-sulfur clusters for their function. Several possible solutions to this problem have been proposed. One is that the first iron-sulfur clusters were synthesized by non-enzymatic processes, such as the reaction of iron and sulfur in the presence of a reducing agent. The non-enzymatic synthesis of iron-sulfur clusters by simple chemical reactions would likely be a very unspecific and inefficient process. Some researchers have proposed that early earth environments, such as hydrothermal vents, would have provided conditions that were conducive to the formation of iron-sulfur clusters through specific mineral catalysis or other mechanisms. These environments would have also provided a source of reducing agents or other reactants that could have facilitated the formation of these clusters in a more controlled manner. While hydrothermal vents and other early earth environments could have provided specific conditions that favored the formation of iron-sulfur clusters, the overall non-specificity of the process would however still be a challenge that cannot be overstated enough. Another claim is that the earliest enzymes that required iron-sulfur clusters for their function were simpler and more primitive than modern enzymes, and were able to operate with simpler or more easily assembled clusters. Over time, these enzymes could have progressed to become more complex and sophisticated and could have developed the ability to synthesize and incorporate more complex iron-sulfur clusters. There is however a large gap between the simpler, more primitive enzymes that would have operated with simpler iron-sulfur clusters, and the highly regulated, multi-step processes involving multiple enzymes that are required for the biosynthesis of iron-sulfur clusters in modern cells. Another hypothesis states that many of the enzymes involved in modern iron-sulfur cluster biosynthesis would have evolved from more ancient enzymes that performed related functions in early life forms. But life was not even extant at such a stage. This is just one tiny problem, among many other factors and processes involved in the emergence of life on Earth, which is unsolved, equally to this problem, and the formation of iron-sulfur clusters is just one piece of a larger puzzle.
The mechanism in Class III RNR enzymes The reaction mechanism of Class III RNR enzymes can be divided into two stages: initiation and propagation.
Initiation:The reaction starts with the binding of a substrate, which is a ribonucleotide, to the enzyme's active site.
The substrate is then converted to a radical species by a radical-generating cofactor, such as adenosylcobalamin or glycyl radical, that is associated with the enzyme.
The radical generated on the substrate abstracts a hydrogen atom from a nearby cysteine residue on the enzyme, forming a cysteine radical and a substrate radical.
Propagation:The substrate radical then undergoes a series of electron and proton transfers within the active site, which leads to the reduction of the substrate to its corresponding deoxyribonucleotide.
The electron and proton transfers involve the participation of several amino acid residues and cofactors that are located within the enzyme's active site.
The cysteine radical generated in the initiation stage is then regenerated back to its original form by a reducing agent, such as thioredoxin or glutaredoxin.
Class III ribonucleotide reductase (RNR) enzymes do not contain iron-sulfur clusters.
Iron-sulfur clusters are cofactors found in Class I and Class II RNR enzymes, which use a different mechanism to catalyze the reduction of ribonucleotides to deoxyribonucleotides. Class III RNR enzymes, on the other hand, use a radical mechanism that involves the participation of radical-generating cofactors, such as adenosylcobalamin or glycyl radical.
It should be noted that not all RNR enzymes contain iron-sulfur clusters. In addition to Class I RNR enzymes, some Class II RNR enzymes also contain iron-sulfur clusters, while others use different cofactors, such as flavodoxin or heme, to generate radicals.
The active site of Class III ribonucleotide reductase (RNR) enzymes
contains several key components that are involved in the reduction of ribonucleotides to deoxyribonucleotides. The following steps outline the general process that occurs in the active site of Class III RNR enzymes:
1. Substrate binding: The ribonucleotide substrate binds to the active site of the enzyme, where it interacts with a radical-generating cofactor, such as adenosylcobalamin or glycyl radical. This interaction results in the generation of a radical on the substrate.
2. Radical transfer: The radical on the substrate undergoes a radical transfer reaction, where it is transferred to a nearby cysteine residue on the enzyme. This forms a cysteine radical and a substrate radical.
3. Propagation: The substrate radical undergoes a series of electron and proton transfers within the active site, which leads to the reduction of the substrate to its corresponding deoxyribonucleotide. This process involves the participation of several amino acid residues and cofactors within the active site, which help to facilitate the transfer of electrons and protons.
4. Cysteine regeneration: The cysteine radical generated in step 2 is then regenerated back to its original form by a reducing agent, such as thioredoxin or glutaredoxin. This allows the enzyme to continue to catalyze the reduction of additional ribonucleotides.
Overall, the active site of Class III RNR enzymes plays a critical role in facilitating the radical mechanism used to reduce ribonucleotides to deoxyribonucleotides. The active site contains specific amino acid residues and cofactors that help to generate and transfer radicals, as well as facilitate the electron and proton transfers necessary for catalysis
AdenosylcobalaminAdenosylcobalamin has one of the most complex structures among all vitamins. It consists of a large, complex molecule known as a corrin ring, which is bound to a central cobalt ion. Cobalt ion is a positively charged ion of the element cobalt. Cobalt is a transition metal that can form different ions depending on its oxidation state. In its ionic form, cobalt can have a +2 or +3 charge. In adenosylcobalamin, the cobalt ion is in the +3 oxidation state and is bound to the corrin ring, forming the core of the coenzyme. The corrin ring is a large, complex organic molecule that is the central part of the adenosylcobalamin coenzyme. It consists of a planar tetrapyrrole ring that contains four nitrogen atoms and is similar in structure to the heme group found in hemoglobin. However, the corrin ring is larger and more complex than the heme group.
The corrin ring has a unique three-dimensional structure that allows it to bind to the cobalt ion at its center and to interact with other molecules during biochemical reactions. It also has several functional groups, including carboxyl and methyl groups, that play important roles in the chemistry of the coenzyme. The corrin ring is synthesized by certain bacteria and archaea, and it is not produced by humans or other animals. It is an essential component of the adenosylcobalamin coenzyme, which is required for several important metabolic processes in the body, including the breakdown of fatty acids and amino acids. Attached to one end of the corrin ring is a nucleotide called adenosine, which serves as a binding site for the enzyme that uses the coenzyme. The other end of the corrin ring is the site where the reaction takes place. The complex structure of adenosylcobalamin is necessary for its ability to serve as a cofactor in a wide range of enzymatic reactions in the body. Its unique structure allows it to act as a "molecular carrier" that can shuttle groups of atoms between different molecules during metabolic processes.
Adenosylcobalamin, also known as coenzyme B12, is a coenzyme that is involved in several important enzymatic reactions in the body. It is a type of cobalamin, which is a group of compounds that contain the metal ion cobalt. Adenosylcobalamin is important for the metabolism of certain amino acids, as well as the breakdown of fatty acids and the synthesis of certain neurotransmitters. It is also involved in the production of energy from glucose through a process called the Krebs cycle. The molecule itself consists of a corrin ring that is coordinated to the cobalt ion. Attached to one end of the corrin ring is a nucleotide called adenosine, which serves as a binding site for the enzyme that uses the coenzyme. The other end of the corrin ring is the site where the reaction takes place. Adenosylcobalamin is produced in the body through a complex biosynthesis pathway that involves several enzymes and cofactors. In this process, cobalt is incorporated into a precursor molecule, which is then modified and processed into the final adenosylcobalamin molecule. Deficiencies in the enzymes involved in this pathway can lead to a range of health problems, including anemia and neurological disorders.






 [/justify]
[/justify]