


Homeostasis in cells, and origin of life scenarios
https://reasonandscience.catsboard.com/t2447-homeostasis-in-cells-and-origin-of-life-scenarios
1. The control of metabolism is a fundamental requirement for all life, with perturbations of metabolic homeostasis underpinning numerous disease-associated pathologies.
2. Any incomplete Metabolic network without the control mechanisms in place to get homeostasis would mean disease and cell death.
3. A minimal metabolic network and the control mechanisms had to be in place from the beginning, which means, and gradualistic explanation of the origin of biological Cells, and life is unrealistic.
Life is an all or nothing business and points to a creative act of God.
One of the key properties of life is Regulation, including homeostasis.
Freeman Dyson, Origins of Life, page 73:
The essential characteristic of living cells is homeostasis, the ability to maintain a steady and more-or-less constant chemical balance in a changing environment. Homeostasis is the machinery of chemical controls and feedback cycles that make sure that each molecular species in a cell is produced in the right proportion, not too much and not too little. Without homeostasis, there can be no ordered metabolism and no quasi-stationary equilibrium deserving the name of life. The question Why is life so complicated? means, in this context, Given that a population of molecules is able to maintain itself in homeostatic equilibrium at a steady level of metabolism, how many different molecular species must the population contain? From the fact that bacteria have generally refused to shrink below a certain level of complexity, we may deduce that this level is in some sense an irreducible minimum. I am conjecturing that the minimum population size required for homeostasis would be ten or twenty thousand monomer units. And more important, I am suggesting that the most promising road to an understanding of the origin of life would be to do experiments like the Spiegelman and Eigen experiments but this time concerned with homeostasis rather than with replication.
The essence of life from the beginning was homeostasis based on a complicated web of molecular structures. Life by its very nature is resistant to simplification, whether on the level of single cells or ecological systems or human societies. Life could tolerate a precisely replicating molecular apparatus only by incorporating it into a translation system that allowed the complexity of the molecular web to be expressed in the form of software. After the transfer of complication from hardware to software, life continued to be a complicated interlocking web in which the replicators were only one component. The replicators were never as firmly in control as Dawkins imagined. In my version the history of life is counterpoint music, a two-part invention with two voices, the voice of the replicators attempting to impose their selfish purposes upon the whole network and the voice of homeostasis tending to maximize diversity of structure and flexibility of function. The tyranny of the replicators was always mitigated by the more ancient cooperative structure of homeostasis that was inherent in every organism.
Homeostasis is the mechanistic fundament of biology, beginning with the protocell 1 Two fundamental properties of life as we recognize it are homeostasis and redox chemistry. Redox homeostasis is defined here as the maintenance of a constant electrochemical potential and ionic concentration gradient across a cellular boundary, despite fluctuations in the electrochemical potential of the external environment and despite changing identities and activities of electron donors and acceptors. 2 The transition to free-living cells depends on the proto-cells’ ability to maintain a proton motive force for energy conversion, providing for metabolism, for active transport, and for synthesis of informational and structural macromolecules. A feature that distinguishes living from non-living matter is homeostasis – the maintenance of a constant internal environment despite changes in the external environment. A second feature of all known life is that living things are composed of spatial compartments, called cells. Cellular homeostasis requires a system of integrated feedback and feedforward, producing adaptive responses to, and anticipation of, ultimately uncontrollable changes in the properties of the outside world.
I have been trying to imagine a framework for the origin of life, guided by a personal philosophy that considers the primal characteristics of life to be homeostasis rather than replication, diversity rather than uniformity, the flexibility of the genome rather than the tyranny of the gene, the error tolerance of the whole rather than the precision of the parts.
A Massachusetts General Hospital (MGH) research team investigating how the earliest stages of life might have developed has discovered a way the first living cells could have met a key challenge -- maintaining a constant internal environment, a process called homeostasis, even when external conditions change. "Modern cells are constantly regulating what they are doing -- synthesizing, degrading and exporting a whole suite of RNAs and proteins -- depending on the cell's particular needs at the time," says Aaron Engelhart, PhD, of the MGH Department of Molecular Biology and the Center for Computational and Integrative Biology, lead author of the paper. "One would expect that the earliest cells weren't nearly as complex as today's cells, but they still had the need to regulate their internal environment. 1
WHAT FUELS LIVING SYTEMS? LIFE AS REDOX CHEMISTRY
In chemical reduction and oxidation, termed redox chemistry for short, one or more electrons move from a donor to an acceptor. In the process, the donor, or reductant, is said to become oxidised, while the acceptor, or oxidant, becomes reduced. A generalised redox reaction involving transfer of a single electron (with its unit of negative charge, –) from molecule A (the electron donor) to molecule B (the electron acceptor). Reading from left to right, we say that A reduces B, while B oxidises A. Free energy is released as the reaction approaches equilibrium. The initial chemical disequilibrium upon which all living things depend for energy arises from the separation of reducing and oxidising chemical species in the first place – it is a redox disequilibrium that allows energy-yielding redox reactions to proceed. This separation results, unaided by life, from geochemical activity, and geochemical gradients across the boundaries of inorganic compartments are now proposed as the earliest, vectorial metabolism of the first living cells, with the inside being more reducing, and the outside more oxidising – a feature maintained by all cells to this day.
Integrating Protein Homeostasis Strategies in Prokaryotes
Abstract
Bacterial cells are frequently exposed to dramatic fluctuations in their environment, which cause perturbation in protein homeostasis and lead to protein misfolding. Bacteria have powerful quality control networks consisting of chaperones and proteases that cooperate to monitor the folding states of proteins and to remove misfolded conformers through either refolding or degradation. The levels of the quality control components are adjusted to the folding state of the cellular proteome through the induction of compartment specific stress responses. In addition, the activities of several quality control components are directly controlled by these stresses, allowing for fast activation. Severe stress can, however, overcome the protective function of the proteostasis network leading to the formation of protein aggregates, which are sequestered at the cell poles. Protein aggregates are either solubilized by AAA+ chaperones or eliminated through cell division, allowing for the generation of damage-free daughter cells.
Unless these quality control mechanisms were in place right from the beginning, would cells not immediately be damaged by environmental changes, and die ?
PROTEIN MISFOLDING AND QUALITY CONTROL SYSTEMS
Although the general principles governing protein folding are similar in all organisms, there are a number of important differences in the folding environments of bacteria and eukaryotic cells. For example, the rate of polypeptide elongation during protein synthesis is significantly faster in bacteria (20 amino acids/sec) compared to eukaryotes (4 amino acids/sec). This difference has an intrinsic impact on cotranslational protein folding. In addition, although some bacteria can form biofilms, many bacteria can flourish unicellularly. As a result, they are more directly exposed to such environmental stresses (e.g., radiation, extreme temperatures or oxidative stress) that could interfere with protein folding, than are multicellular organisms.
To ensure protein homeostasis, bacteria have sophisticated quality control systems consisting primarily of chaperones and proteases that exert multiple activities, which can be roughly divided into the following categories:
(1) de novo folding of newly synthesized proteins
(2) preventing aggregation of unfolded proteins
(3) removing of terminally misfolded proteins by degradation
(4) resolubilizing protein aggregates for subsequent refolding or degradation
we describe the prokaryotic cytosolic proteostasis network with a focus on Escherichia coli. However, because of the conserved nature of the proteostatic network, many of the principles derived from studies of E. coli can be applied to other bacteria.
RIBOSOME-BOUND TRIGGER FACTOR WELCOMES NASCENT POLYPEPTIDE CHAINS
Nascent polypeptides emerge from the polypeptide exit tunnel of the ribosome in a more-or-less unfolded conformation. Consequently, nascent proteins could populate partially folded, aggregation-prone states that require the cotranslational assistance of chaperones to fold correctly. The ribosome-associated chaperone trigger factor (TF) interacts with nascent proteins to promote efficient de novo protein folding
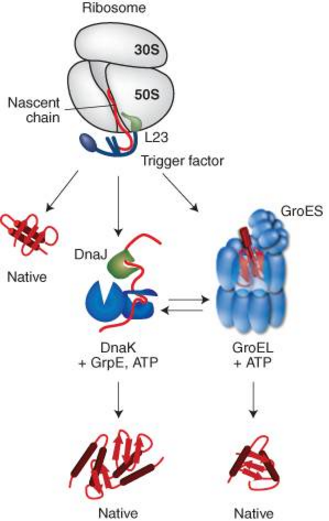
Figure 1. Interplay of chaperone system during de novo protein folding in E. coli.
Nascent polypeptides initially interact with ribosome-bound Trigger Factor (TF), which binds to the ribosomal protein L23. On release from TF, newly synthesized proteins either fold spontaneously (roughly estimated two thirds of cytosolic proteins under physiological growth conditions) or require further folding-assistance by downstream chaperones, namely the Hsp70 chaperone DnaK, which acts together with its co-chaperone DnaJ and the nucleotide exchange factor GrpE, and/or the Hsp60 chaperone GroEL with its co-chaperone GroES. The ATP-dependent DnaK- and GroEL-machineries may act co- and/or posttranslationally.
ADJUSTING QUALITY CONTROL NETWORKS TO ENVIRONMENTAL STRESS: REGULATION OF STRESS RESPONSES IN BACTERIA
As a result of their lifestyle, unicellular prokaryotes (and to a lesser extent, those growing in biofilms) are often exposed to dramatic fluctuations in their environment, which can result in a perturbation of protein homeostasis. For example, the enteric commensals of mammals, such as E. coli, must survive a sudden shift from ambient to body temperature (∼36°C–40°C) at ingestion, acid shock in the stomach (∼pH 1–2), and a return to alkaline pH (∼pH 8–9) in the small intestine to successfully colonize a new host. Free-living organisms, which are more directly exposed to environmental fluctuations, must often survive even harsher folding stresses. These stresses not only disrupt the folding of newly synthesized proteins but can also cause misfolding of already folded proteins. Bacteria have therefore sophisticated stress responses that can react to such threats to proteostasis through the induction of chaperones and proteases.
The best-studied folding stress, heat, induces the expression of chaperone and protease genes by various regulatory circuits, which work together to fine-tune the stress response. These circuits can be grouped into two general classes: (i) temperature-responsive mRNA and thermolabile transcriptional regulators that directly respond to temperature changes and (ii) transcriptional regulators that are controlled by chaperones or proteases, which indirectly monitor the general folding state of the cell.
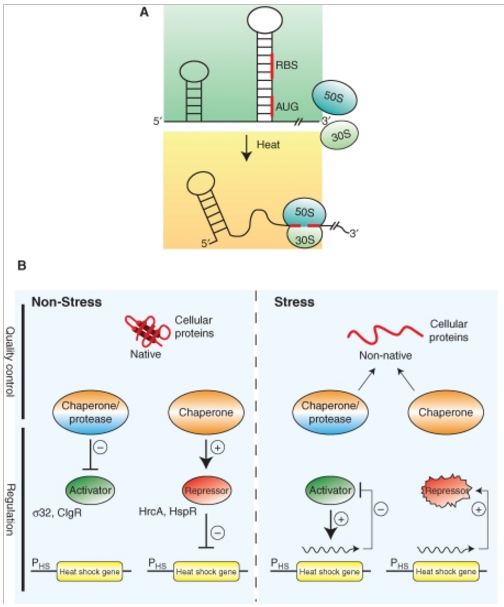
Figure 2. Regulation of bacterial stress responses.
(A) Principle of RNA thermometers. At low temperatures, the ribosomal binding site (RBS) and the AUG start codon of an mRNA encoding for a stress gene is base paired and not accessible. On heat shock, the structure around the RBS melts allowing for ribosome binding (30S and 50S) and translation.
(B) Chaperones and proteases link the cellular folding state to stress gene expression. Under nonstress conditions, expression of stress genes is inhibited through (1) inhibition of transcriptional activators by chaperones or proteases that either sequester the regulators or degrade them or (2) repressor proteins that require chaperone assistance for activity. During environmental stress misfolded proteins accumulate, which titrate chaperones and proteases from their regulatory roles and resulting in the expression of stress genes through (1) release or stabilization of transcriptional activators or (2) inactivation of repressor proteins. Expression of stress genes initiates an inactivation feedback loop restoring nonstress gene expression.
CHALLENGES TO THE QUALITY CONTROL SYSTEMS DURING STRESS CONDITIONS
Exposure of cells to physical and chemical denaturants, such as increased temperature, changes in ionic strength, oxidative stress or the presence of heavy metals, can disturb proteostasis and lead to the accumulation of misfolded proteins, which are at risk of aggregation. Here, we have concentrated on heat stress, because it is the best-characterized stress condition.
The primary strategy of cellular quality control systems during heat stress is to keep client proteins in a soluble, folding-competent state. Although most chaperone classes have been shown to prevent the aggregation of heat-labile reporters in vitro, they clearly have different functional roles during heat stress in vivo. In E. coli, the DnaK system is most important in preventing the aggregation of misfolded proteins during heat stress (Fig. 3) (Gragerov et al. 1992; Mogk et al. 1999).
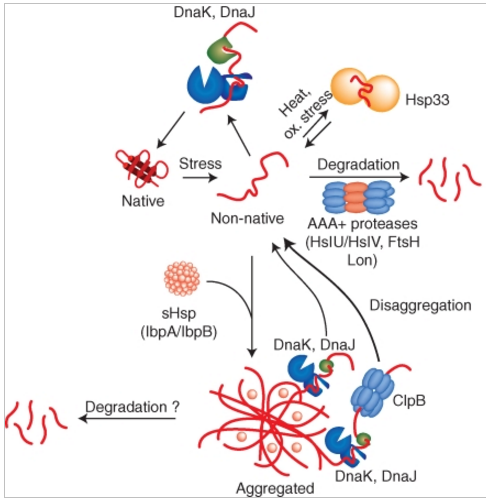
Figure 3. Activities of bacterial quality control systems during environmental stress.
Environmental stress like heat shock can cause protein-unfolding leading to the accumulation of misfolded protein species. Misfolded proteins are either refolded by the DnaK chaperone and its co-chaperone DnaJ or are removed by AAA+ proteases including e.g., Lon, ClpC/ClpP or HslU/HslV. The holding chaperone Hsp33 becomes important under oxidative and thermal stress and prevents protein aggregation. Severe stress conditions can overburden the protective capacity of quality control systems causing protein aggregation. sHsps coaggregate with misfolded protein species thereby changing the architecture (physical properties) of aggregates and allowing for more efficient protein disaggregation by chaperones. DnaK/DnaJ in cooperation with the AAA+ chaperone ClpB efficiently solubilize protein aggregates by extracting single unfolded protein species, whereas DnaK/DnaJ alone have limited disaggregation capacity. AAA+ proteases (ClpC/ClpP) might also act on aggregated protein species.
This role is based on its promiscuous and efficient substrate binding capacity and its high cellular concentration. Accordingly, dnaK mutants show a temperature-sensitive growth phenotype and strongly increased protein aggregation on heat shock. Heat shock temperatures also directly modulate the activity of the DnaK system by causing reversible inactivation of the nucleotide exchange factor GrpE (Groemping and Reinstein 2001). Inactivation of GrpE largely retards substrate release by slowing down ATP/ADP exchange, which freezes DnaK in the high affinity state for substrates. In this sense GrpE acts as a thermosensor and its activity control ensures that substrates remain bound to DnaK during stress conditions.
REMOVAL OF MISFOLDED PROTEINS BY AAA+ PROTEASES
Often, the cell cannot salvage misfolded proteins by the aforementioned chaperone systems. In such cases, prokaryotic cells protect themselves from the potentially detrimental effects of these terminally misfolded protein species by degrading them using AAA+ proteases (Fig. 3). AAA+ proteases are compartmentalized proteases that consist of two functional units with separate activities: a barrel-shaped proteolytic subunit (e.g., heptameric ClpP or hexameric HslV) and a hexameric member of the AAA+ family of ATPases (e.g., ClpA, ClpC, HslU), which act as chaperones to unfold the protein substrate and feed it into the central proteolytic channel of the protease (Sauer et al. 2004). A number of AAA+ proteases are present in the cytoplasm of E. coli, in which the protease and AAA+ subunits may be encoded in one (e.g., Lon and FtsH), or two (e.g., ClpA/ClpP, ClpX/ClpP, and HslU/HslV) polypeptide chains.
Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines
To maintain protein homeostasis, AAA+ proteolytic machines degrade damaged and unneeded proteins in bacteria, archaea and eukaryotes. This process involves the ATP-dependent unfolding of a target protein and its subsequent translocation into a self-compartmentalized proteolytic chamber. Related AAA+ enzymes also disaggregate and remodel proteins. Recent structural and biochemical studies, in combination with direct visualization of unfolding and translocation in single-molecule experiments, have illuminated the molecular mechanisms behind these processes and suggest how remodelling of macromolecular complexes by AAA+ enzymes could occur without global denaturation. In this Review, we discuss the structural and mechanistic features of AAA+ proteases and remodelling machines, focusing on the bacterial ClpXP and ClpX as paradigms.
Sculpting the Proteome with AAA+ Proteases and Disassembly Machines
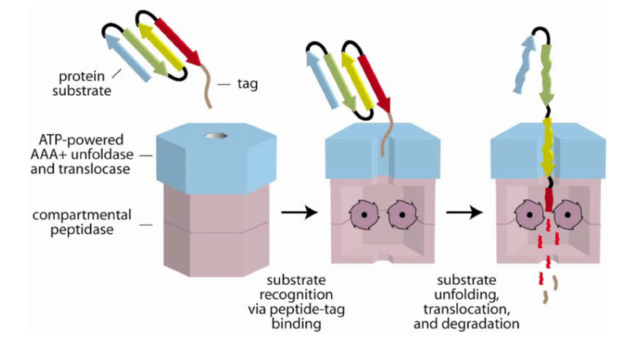
Cartoon Model of Substrate Recognition and Degradation by a AAA+ Protease
The recognition step is mediated by binding of a peptide tag (brown) on the protein substrate to a AAA+ ATPase (blue). In subsequent steps, the protein is unfolded, translocated into a compartmental peptidase (magenta), and degraded. Peptide fragments are shown diffusing out of the peptidase, but active participation of the ATPase may be required for exit of large fragments.
The mechanistic principle of substrate selection in general proteolysis by AAA+ proteases has been best characterized for Lon, which plays an important role in the removal of premature translational termination products and proteins containing nonnatural amino acids. The recognition of misfolded proteins seems to be largely mediated by the amino-terminal domain of Lon, as respective Lon deletion variants are impaired in the degradation of casein . Lon recognizes hydrophobic peptide stretches that are enriched in aromatic residues, which are typically buried in the structure of the folded protein and allows the selective identification of misfolded proteins.
Some misfolded model substrates are stabilized in cells lacking specific chaperone systems, which suggest that AAA+ proteases work together with chaperones in protein degradation. However, the interplay between chaperones and proteases in determining the fate of a misfolded protein (i.e., refolding v. degradation) is not well understood. One possibility is that substrates are kinetically partitioned such that proteins that do not fold rapidly enough are degraded. Different misfolded protein substrates could display different affinities for proteases and chaperones. For example, although both DnaK and Lon recognize hydrophobic sequences, hydrophobic clusters lacking aromatic residues are poor Lon substrates whereas the interaction of DnaK with a substrate is less dependent on the presence of aromatic amino acids. It is also possible that chaperones alter the conformation of substrates, rendering them more accessible for proteolysis. One example is σ32, which requires the activity of the DnaK system for efficient degradation by the AAA+ protease FtsH. DnaK and DnaJ binding induces conformational changes within the σ32 polypeptide, which may be the prerequisite for recognition by FtsH . However, in most cases, chaperones and proteases seem to compete for binding to misfolded protein species and their different activities–refolding vs. degradation—can at least partially compensate for the loss of each other. Thus, the AAA+ proteases ClpX/ClpP and Lon become essential for E. coli viability at high temperatures only if the levels of DnaK are reduced.
1. http://www.news-medical.net/news/20160317/Homeostasis-may-have-allowed-first-living-cells-to-maintain-internal-environment.aspx
https://reasonandscience.catsboard.com/t2447-homeostasis-in-cells-and-origin-of-life-scenarios
1. The control of metabolism is a fundamental requirement for all life, with perturbations of metabolic homeostasis underpinning numerous disease-associated pathologies.
2. Any incomplete Metabolic network without the control mechanisms in place to get homeostasis would mean disease and cell death.
3. A minimal metabolic network and the control mechanisms had to be in place from the beginning, which means, and gradualistic explanation of the origin of biological Cells, and life is unrealistic.
Life is an all or nothing business and points to a creative act of God.
One of the key properties of life is Regulation, including homeostasis.
Freeman Dyson, Origins of Life, page 73:
The essential characteristic of living cells is homeostasis, the ability to maintain a steady and more-or-less constant chemical balance in a changing environment. Homeostasis is the machinery of chemical controls and feedback cycles that make sure that each molecular species in a cell is produced in the right proportion, not too much and not too little. Without homeostasis, there can be no ordered metabolism and no quasi-stationary equilibrium deserving the name of life. The question Why is life so complicated? means, in this context, Given that a population of molecules is able to maintain itself in homeostatic equilibrium at a steady level of metabolism, how many different molecular species must the population contain? From the fact that bacteria have generally refused to shrink below a certain level of complexity, we may deduce that this level is in some sense an irreducible minimum. I am conjecturing that the minimum population size required for homeostasis would be ten or twenty thousand monomer units. And more important, I am suggesting that the most promising road to an understanding of the origin of life would be to do experiments like the Spiegelman and Eigen experiments but this time concerned with homeostasis rather than with replication.
The essence of life from the beginning was homeostasis based on a complicated web of molecular structures. Life by its very nature is resistant to simplification, whether on the level of single cells or ecological systems or human societies. Life could tolerate a precisely replicating molecular apparatus only by incorporating it into a translation system that allowed the complexity of the molecular web to be expressed in the form of software. After the transfer of complication from hardware to software, life continued to be a complicated interlocking web in which the replicators were only one component. The replicators were never as firmly in control as Dawkins imagined. In my version the history of life is counterpoint music, a two-part invention with two voices, the voice of the replicators attempting to impose their selfish purposes upon the whole network and the voice of homeostasis tending to maximize diversity of structure and flexibility of function. The tyranny of the replicators was always mitigated by the more ancient cooperative structure of homeostasis that was inherent in every organism.
Homeostasis is the mechanistic fundament of biology, beginning with the protocell 1 Two fundamental properties of life as we recognize it are homeostasis and redox chemistry. Redox homeostasis is defined here as the maintenance of a constant electrochemical potential and ionic concentration gradient across a cellular boundary, despite fluctuations in the electrochemical potential of the external environment and despite changing identities and activities of electron donors and acceptors. 2 The transition to free-living cells depends on the proto-cells’ ability to maintain a proton motive force for energy conversion, providing for metabolism, for active transport, and for synthesis of informational and structural macromolecules. A feature that distinguishes living from non-living matter is homeostasis – the maintenance of a constant internal environment despite changes in the external environment. A second feature of all known life is that living things are composed of spatial compartments, called cells. Cellular homeostasis requires a system of integrated feedback and feedforward, producing adaptive responses to, and anticipation of, ultimately uncontrollable changes in the properties of the outside world.
I have been trying to imagine a framework for the origin of life, guided by a personal philosophy that considers the primal characteristics of life to be homeostasis rather than replication, diversity rather than uniformity, the flexibility of the genome rather than the tyranny of the gene, the error tolerance of the whole rather than the precision of the parts.
A Massachusetts General Hospital (MGH) research team investigating how the earliest stages of life might have developed has discovered a way the first living cells could have met a key challenge -- maintaining a constant internal environment, a process called homeostasis, even when external conditions change. "Modern cells are constantly regulating what they are doing -- synthesizing, degrading and exporting a whole suite of RNAs and proteins -- depending on the cell's particular needs at the time," says Aaron Engelhart, PhD, of the MGH Department of Molecular Biology and the Center for Computational and Integrative Biology, lead author of the paper. "One would expect that the earliest cells weren't nearly as complex as today's cells, but they still had the need to regulate their internal environment. 1
WHAT FUELS LIVING SYTEMS? LIFE AS REDOX CHEMISTRY
In chemical reduction and oxidation, termed redox chemistry for short, one or more electrons move from a donor to an acceptor. In the process, the donor, or reductant, is said to become oxidised, while the acceptor, or oxidant, becomes reduced. A generalised redox reaction involving transfer of a single electron (with its unit of negative charge, –) from molecule A (the electron donor) to molecule B (the electron acceptor). Reading from left to right, we say that A reduces B, while B oxidises A. Free energy is released as the reaction approaches equilibrium. The initial chemical disequilibrium upon which all living things depend for energy arises from the separation of reducing and oxidising chemical species in the first place – it is a redox disequilibrium that allows energy-yielding redox reactions to proceed. This separation results, unaided by life, from geochemical activity, and geochemical gradients across the boundaries of inorganic compartments are now proposed as the earliest, vectorial metabolism of the first living cells, with the inside being more reducing, and the outside more oxidising – a feature maintained by all cells to this day.
Integrating Protein Homeostasis Strategies in Prokaryotes
Abstract
Bacterial cells are frequently exposed to dramatic fluctuations in their environment, which cause perturbation in protein homeostasis and lead to protein misfolding. Bacteria have powerful quality control networks consisting of chaperones and proteases that cooperate to monitor the folding states of proteins and to remove misfolded conformers through either refolding or degradation. The levels of the quality control components are adjusted to the folding state of the cellular proteome through the induction of compartment specific stress responses. In addition, the activities of several quality control components are directly controlled by these stresses, allowing for fast activation. Severe stress can, however, overcome the protective function of the proteostasis network leading to the formation of protein aggregates, which are sequestered at the cell poles. Protein aggregates are either solubilized by AAA+ chaperones or eliminated through cell division, allowing for the generation of damage-free daughter cells.
Unless these quality control mechanisms were in place right from the beginning, would cells not immediately be damaged by environmental changes, and die ?
PROTEIN MISFOLDING AND QUALITY CONTROL SYSTEMS
Although the general principles governing protein folding are similar in all organisms, there are a number of important differences in the folding environments of bacteria and eukaryotic cells. For example, the rate of polypeptide elongation during protein synthesis is significantly faster in bacteria (20 amino acids/sec) compared to eukaryotes (4 amino acids/sec). This difference has an intrinsic impact on cotranslational protein folding. In addition, although some bacteria can form biofilms, many bacteria can flourish unicellularly. As a result, they are more directly exposed to such environmental stresses (e.g., radiation, extreme temperatures or oxidative stress) that could interfere with protein folding, than are multicellular organisms.
To ensure protein homeostasis, bacteria have sophisticated quality control systems consisting primarily of chaperones and proteases that exert multiple activities, which can be roughly divided into the following categories:
(1) de novo folding of newly synthesized proteins
(2) preventing aggregation of unfolded proteins
(3) removing of terminally misfolded proteins by degradation
(4) resolubilizing protein aggregates for subsequent refolding or degradation
we describe the prokaryotic cytosolic proteostasis network with a focus on Escherichia coli. However, because of the conserved nature of the proteostatic network, many of the principles derived from studies of E. coli can be applied to other bacteria.
RIBOSOME-BOUND TRIGGER FACTOR WELCOMES NASCENT POLYPEPTIDE CHAINS
Nascent polypeptides emerge from the polypeptide exit tunnel of the ribosome in a more-or-less unfolded conformation. Consequently, nascent proteins could populate partially folded, aggregation-prone states that require the cotranslational assistance of chaperones to fold correctly. The ribosome-associated chaperone trigger factor (TF) interacts with nascent proteins to promote efficient de novo protein folding
Figure 1. Interplay of chaperone system during de novo protein folding in E. coli.
Nascent polypeptides initially interact with ribosome-bound Trigger Factor (TF), which binds to the ribosomal protein L23. On release from TF, newly synthesized proteins either fold spontaneously (roughly estimated two thirds of cytosolic proteins under physiological growth conditions) or require further folding-assistance by downstream chaperones, namely the Hsp70 chaperone DnaK, which acts together with its co-chaperone DnaJ and the nucleotide exchange factor GrpE, and/or the Hsp60 chaperone GroEL with its co-chaperone GroES. The ATP-dependent DnaK- and GroEL-machineries may act co- and/or posttranslationally.
ADJUSTING QUALITY CONTROL NETWORKS TO ENVIRONMENTAL STRESS: REGULATION OF STRESS RESPONSES IN BACTERIA
As a result of their lifestyle, unicellular prokaryotes (and to a lesser extent, those growing in biofilms) are often exposed to dramatic fluctuations in their environment, which can result in a perturbation of protein homeostasis. For example, the enteric commensals of mammals, such as E. coli, must survive a sudden shift from ambient to body temperature (∼36°C–40°C) at ingestion, acid shock in the stomach (∼pH 1–2), and a return to alkaline pH (∼pH 8–9) in the small intestine to successfully colonize a new host. Free-living organisms, which are more directly exposed to environmental fluctuations, must often survive even harsher folding stresses. These stresses not only disrupt the folding of newly synthesized proteins but can also cause misfolding of already folded proteins. Bacteria have therefore sophisticated stress responses that can react to such threats to proteostasis through the induction of chaperones and proteases.
The best-studied folding stress, heat, induces the expression of chaperone and protease genes by various regulatory circuits, which work together to fine-tune the stress response. These circuits can be grouped into two general classes: (i) temperature-responsive mRNA and thermolabile transcriptional regulators that directly respond to temperature changes and (ii) transcriptional regulators that are controlled by chaperones or proteases, which indirectly monitor the general folding state of the cell.
Figure 2. Regulation of bacterial stress responses.
(A) Principle of RNA thermometers. At low temperatures, the ribosomal binding site (RBS) and the AUG start codon of an mRNA encoding for a stress gene is base paired and not accessible. On heat shock, the structure around the RBS melts allowing for ribosome binding (30S and 50S) and translation.
(B) Chaperones and proteases link the cellular folding state to stress gene expression. Under nonstress conditions, expression of stress genes is inhibited through (1) inhibition of transcriptional activators by chaperones or proteases that either sequester the regulators or degrade them or (2) repressor proteins that require chaperone assistance for activity. During environmental stress misfolded proteins accumulate, which titrate chaperones and proteases from their regulatory roles and resulting in the expression of stress genes through (1) release or stabilization of transcriptional activators or (2) inactivation of repressor proteins. Expression of stress genes initiates an inactivation feedback loop restoring nonstress gene expression.
CHALLENGES TO THE QUALITY CONTROL SYSTEMS DURING STRESS CONDITIONS
Exposure of cells to physical and chemical denaturants, such as increased temperature, changes in ionic strength, oxidative stress or the presence of heavy metals, can disturb proteostasis and lead to the accumulation of misfolded proteins, which are at risk of aggregation. Here, we have concentrated on heat stress, because it is the best-characterized stress condition.
The primary strategy of cellular quality control systems during heat stress is to keep client proteins in a soluble, folding-competent state. Although most chaperone classes have been shown to prevent the aggregation of heat-labile reporters in vitro, they clearly have different functional roles during heat stress in vivo. In E. coli, the DnaK system is most important in preventing the aggregation of misfolded proteins during heat stress (Fig. 3) (Gragerov et al. 1992; Mogk et al. 1999).
Figure 3. Activities of bacterial quality control systems during environmental stress.
Environmental stress like heat shock can cause protein-unfolding leading to the accumulation of misfolded protein species. Misfolded proteins are either refolded by the DnaK chaperone and its co-chaperone DnaJ or are removed by AAA+ proteases including e.g., Lon, ClpC/ClpP or HslU/HslV. The holding chaperone Hsp33 becomes important under oxidative and thermal stress and prevents protein aggregation. Severe stress conditions can overburden the protective capacity of quality control systems causing protein aggregation. sHsps coaggregate with misfolded protein species thereby changing the architecture (physical properties) of aggregates and allowing for more efficient protein disaggregation by chaperones. DnaK/DnaJ in cooperation with the AAA+ chaperone ClpB efficiently solubilize protein aggregates by extracting single unfolded protein species, whereas DnaK/DnaJ alone have limited disaggregation capacity. AAA+ proteases (ClpC/ClpP) might also act on aggregated protein species.
This role is based on its promiscuous and efficient substrate binding capacity and its high cellular concentration. Accordingly, dnaK mutants show a temperature-sensitive growth phenotype and strongly increased protein aggregation on heat shock. Heat shock temperatures also directly modulate the activity of the DnaK system by causing reversible inactivation of the nucleotide exchange factor GrpE (Groemping and Reinstein 2001). Inactivation of GrpE largely retards substrate release by slowing down ATP/ADP exchange, which freezes DnaK in the high affinity state for substrates. In this sense GrpE acts as a thermosensor and its activity control ensures that substrates remain bound to DnaK during stress conditions.
REMOVAL OF MISFOLDED PROTEINS BY AAA+ PROTEASES
Often, the cell cannot salvage misfolded proteins by the aforementioned chaperone systems. In such cases, prokaryotic cells protect themselves from the potentially detrimental effects of these terminally misfolded protein species by degrading them using AAA+ proteases (Fig. 3). AAA+ proteases are compartmentalized proteases that consist of two functional units with separate activities: a barrel-shaped proteolytic subunit (e.g., heptameric ClpP or hexameric HslV) and a hexameric member of the AAA+ family of ATPases (e.g., ClpA, ClpC, HslU), which act as chaperones to unfold the protein substrate and feed it into the central proteolytic channel of the protease (Sauer et al. 2004). A number of AAA+ proteases are present in the cytoplasm of E. coli, in which the protease and AAA+ subunits may be encoded in one (e.g., Lon and FtsH), or two (e.g., ClpA/ClpP, ClpX/ClpP, and HslU/HslV) polypeptide chains.
Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines
To maintain protein homeostasis, AAA+ proteolytic machines degrade damaged and unneeded proteins in bacteria, archaea and eukaryotes. This process involves the ATP-dependent unfolding of a target protein and its subsequent translocation into a self-compartmentalized proteolytic chamber. Related AAA+ enzymes also disaggregate and remodel proteins. Recent structural and biochemical studies, in combination with direct visualization of unfolding and translocation in single-molecule experiments, have illuminated the molecular mechanisms behind these processes and suggest how remodelling of macromolecular complexes by AAA+ enzymes could occur without global denaturation. In this Review, we discuss the structural and mechanistic features of AAA+ proteases and remodelling machines, focusing on the bacterial ClpXP and ClpX as paradigms.
Sculpting the Proteome with AAA+ Proteases and Disassembly Machines
Cartoon Model of Substrate Recognition and Degradation by a AAA+ Protease
The recognition step is mediated by binding of a peptide tag (brown) on the protein substrate to a AAA+ ATPase (blue). In subsequent steps, the protein is unfolded, translocated into a compartmental peptidase (magenta), and degraded. Peptide fragments are shown diffusing out of the peptidase, but active participation of the ATPase may be required for exit of large fragments.
The mechanistic principle of substrate selection in general proteolysis by AAA+ proteases has been best characterized for Lon, which plays an important role in the removal of premature translational termination products and proteins containing nonnatural amino acids. The recognition of misfolded proteins seems to be largely mediated by the amino-terminal domain of Lon, as respective Lon deletion variants are impaired in the degradation of casein . Lon recognizes hydrophobic peptide stretches that are enriched in aromatic residues, which are typically buried in the structure of the folded protein and allows the selective identification of misfolded proteins.
Some misfolded model substrates are stabilized in cells lacking specific chaperone systems, which suggest that AAA+ proteases work together with chaperones in protein degradation. However, the interplay between chaperones and proteases in determining the fate of a misfolded protein (i.e., refolding v. degradation) is not well understood. One possibility is that substrates are kinetically partitioned such that proteins that do not fold rapidly enough are degraded. Different misfolded protein substrates could display different affinities for proteases and chaperones. For example, although both DnaK and Lon recognize hydrophobic sequences, hydrophobic clusters lacking aromatic residues are poor Lon substrates whereas the interaction of DnaK with a substrate is less dependent on the presence of aromatic amino acids. It is also possible that chaperones alter the conformation of substrates, rendering them more accessible for proteolysis. One example is σ32, which requires the activity of the DnaK system for efficient degradation by the AAA+ protease FtsH. DnaK and DnaJ binding induces conformational changes within the σ32 polypeptide, which may be the prerequisite for recognition by FtsH . However, in most cases, chaperones and proteases seem to compete for binding to misfolded protein species and their different activities–refolding vs. degradation—can at least partially compensate for the loss of each other. Thus, the AAA+ proteases ClpX/ClpP and Lon become essential for E. coli viability at high temperatures only if the levels of DnaK are reduced.
1. http://www.news-medical.net/news/20160317/Homeostasis-may-have-allowed-first-living-cells-to-maintain-internal-environment.aspx