Chapter 8The origin of VirusesPeter Pollard (2014): Viruses are vital to our very existence. No one seems to stick up for the good guys that keep ecosystems diverse and balanced. Phage species richness is immense with a predicted 100 million phage species.
The sheer number of these good viruses is astonishing. Their concentration in a productive lake or river is often 100 million per millilitre – that’s more than four times the population of Australia squeezed into a ¼ of a teaspoon of water. Globally the oceans contain 10^30 viruses.
Curtis A. Suttle (2005): If the viruses were stretched end to end they would span ~10 million light years. In context, this is equivalent to the carbon in ~75 million blue whales (~10% carbon, by weight), and is ~100 times the distance across our own galaxy. This makes viruses the most abundant biological entity in the water column of the world’s oceans, and the second largest component of biomass after prokaryotes. 1
Polland continues: What are viruses? Viruses are not living organisms. They are simply bits of genetic material (DNA or RNA) covered in protein, that behave like parasites. They attach to their target cell (the host), inject their genetic material, and replicate themselves using the host cells’ metabolic pathways, as you can see in the figure below. Then the new viruses break out of the cell — the cell explodes (lyses), releasing hundreds of viruses. Viruses are very picky about who they will infect. Each viral type has evolved to infect only one host species. Viruses that infect bacteria dominate our world. A virus that infects one species of bacteria won’t infect another bacterial species, and definitely can’t infect you. We have our own suite of a couple of dozen viral types that cause us disease and death. Algae and plants are primary producers, and the foundation of the world’s ecosystems. Using sunlight they turn raw elements like carbon dioxide, nitrogen and phosphorus into organic matter. In turn, they are eaten by herbivores, which are in turn eaten by other animals, and so on. Energy and nutrients are passed on up the food chain until animals die. But what ensures that the primary producers get the raw elements they need to get started? The answer hinges on the viruses’ relationship with bacteria. A virus doesn’t go hunting for its prey. It relies on randomly encountering a host — it’s a numbers game. When the host, such as a bacterial cell, grows rapidly, that number increases. The more of a bacterial species there is, the more likely it will come into contact with its viral nemesis — “killing the winner”. This means that no single bacterial species dominates an ecosystem for very long. In freshwater, for example, you see very high rates of bacterial growth. You would think this high bacterial production would become part of the food chain and end up as fish food. But that is rarely the case. We now realise that the bacteria actually disappear from these ecosystems. So where do the bacteria go? The answer lies in the interaction between bacteria and viruses. When a virus bursts open a bacterial cell its “guts” are spewed back into the water along with all the new viruses. The cell contents then become food for the neighboring bacteria, thereby stimulating their growth. These bacteria increase in numbers and upon coming into contact with their viral nemesis they, too, become infected and lyse. This process of viral infection, lysis, and nutrient release occur over and over again. Bacteria are, in effect, cannibalizing each other with the help of their associated viruses. Very quickly, the elements that support the food web are put back into circulation with the help of viruses.
It’s the combination of high bacterial growth and viral infection that keeps ecosystems functioning. Thus viruses are a critical part of inorganic nutrient recycling. So while they are tiny and seem insignificant, viruses actually play an essential global role in the recycling of nutrients through food webs.
The origin of viruses, essential agents for life, is another mystery besides the origin of life This is a major conundrum. Researchers struggle to find a coherent narrative to explain the fact that life depends on Viruses and vice versa. If we want to elucidate how life began, it is important to give a closer look as well to the origin of Viruses. Viruses are rarely in the spotlight when it comes to elucidating biological origins. Unjustifiably so, since they are essential for life.
What came first, cells or viruses? This is a classical chicken & egg problem: Gladys Kostyrka (2016): Cells depend on viruses, but viruses depend on cells as a host for replication. What came first? How could viruses play critical roles in the OL if life relies on cellular organization and if viruses are defined as parasites of cells? In other words, how could viruses play a role in the emergence of cellular life if the existence of cells is a prerequisite for the existence of viruses? 2
Colin Hill (2021): From the giant, ameba-infecting marine viruses to the tiny Porcine circovirus harboring only two genes, viruses and their cellular hosts are ecologically and evolutionarily intertwined. 3
C.Arnold (2014): Koonin believes in a Virus World. According to him, the ancestors of modern viruses emerged when all life was still a floating stew of genetic information, amino acids, and lipids. The earliest pieces of genetic material were, according to him, likely short pieces of RNA with relatively few genes that often parasitized other floating bits of genetic material to make copies of themselves. These naked pieces of genetic information swapped genes at a primeval genetic flea market, appropriating hand-me-downs from other elements and discarding genes that were no longer needed.
This appears to be one of the many attempts to make sense and explain the origin of viruses, but it is not convincing. Recent evidence puts a question mark on such hypotheses. C.Arnold continues: The largest virus ever discovered, pithovirus is more massive than even some bacteria. Most viruses copy themselves by hijacking their host's molecular machinery. But pithovirus is much more independent, possessing some replication machinery of its own. Pithovirus's relatively large number of genes also differentiated it from other viruses, which are often genetically simple—the smallest have a mere four genes. Pithovirus has around 500 genes, and some are used for complex tasks such as making proteins and repairing and replicating DNA. "It was so different from what we were taught about viruses," Abergel said. (Also see "Virus-Infecting Virus Fuels Definition of Life Debate.") The stunning find, first revealed in 2014, isn't just expanding scientists' notions of what a virus can be. It is reframing the debate over the origins of life. The ancestors of modern viruses are far from being evolutionary laggards.
The origin of their complexity demands as much an explanation as the origin of the first cells. C.Arnold again: The predominant theories for the origin of viruses propose that they emerged either from a type of degenerate cell that had lost the ability to replicate on its own or from genes that had escaped their cellular confines. Giant viruses, first described in 2003, began to change that line of thinking for some scientists. These novel entities represented an entirely new kind of virus. Indeed, the first specimen—isolated from an amoeba living in a cooling tower in England—was so odd that it took scientists years to understand what they had. The scientists named it mimivirus, for MImicking MIcrobe virus, because amoebae appear to mistake it for their typical bacterial meal. Then, next, with a staggeringly high number of genes, approximately 2,500, pandoravirus seemed to herald an entirely new class of viral life. "More than 90 percent of its genes do not resemble anything else found on Earth.
If viruses developed from cells, they should be less diverse because cells would contain the entire range of genes available to viruses. Viruses are also more diverse when it comes to reproduction. "Cells only have two main ways of replicating their DNA. Viruses, on the other hand, have many more methods at their disposal. 4
Hugh Ross (2020): Without viruses, bacteria would multiply and, within a relatively short time period, occupy every niche and cranny on Earth’s surface. The planet would become a giant bacterial slime ball. Those sextillions of bacteria would consume all the resources essential for life and die. Viruses keep Earth’s bacterial population in check. They break up and kill bacteria at the just-right rates and in the just-right locations so as to maintain a population and diversity of bacteria that is optimal for both the bacteria and all the other life forms. It is important to note that all multicellular life depends on bacteria being present at the optimal population level and optimal diversity. We wouldn’t be here without viruses! Viruses also play a crucial role in Earth’s carbon cycle. They and the bacterial fragments they create are carbonaceous substances. Through their role in precipitation, they collect as vast carbonaceous sheets on the surfaces of the world’s oceans. These sheets or mats of viruses and bacterial fragments sink slowly and eventually land on the ocean floors. As they are sinking they provide important nutrients for deep-sea and benthic (bottom-dwelling) life. Plate tectonics drive much of the viral and bacterial fragments into Earth’s crust and mantle where some of that carbonaceous material is returned to the atmosphere through volcanic eruptions.5 Virus-archaea interactions play a central role in global biogeochemical cycles. Ramesh K Goel (2021): Viruses play vital biogeochemical and ecological roles by (a) expressing auxiliary metabolic genes during infection, (b) enhancing the lateral transfer of host genes, and (c) inducing host mortality. Even in harsh and extreme environments, viruses are major players in carbon and nutrient recycling from organic matter. 6 Eugene V. Koonin (2020): Lytic infections (involving the replication of a viral genome) of cellular organisms, primarily bacteria, by viruses play a central role in the biological matter turnover in the biosphere. Considering the enormous abundance and diversity of viruses and other mobile genetic elements (MGEs), and the ubiquitous interactions between MGEs and cellular hosts, a thorough investigation of the evolutionary relationships among viruses and mobile genetic elements (MGEs) is essential to advance our understanding of the evolution of life 7 Eugene V Koonin (2013): Virus killing of marine bacteria and protists largely determines the composition of the biota, provides a major source of organic matter for consumption by heterotrophic organisms, and also defines the formation of marine sediments through the deposition of skeletons of killed plankton organisms such as foraminifera and diatoms. 8 Rachel Nuwer (2020): If all viruses suddenly disappeared, the world would be a wonderful place for about a day and a half, and then we’d all die – that’s the bottom line. The vast majority of viruses are not pathogenic to humans, and many play integral roles in propping up ecosystems. Others maintain the health of individual organisms – everything from fungi and plants to insects and humans. “We live in a balance, in a perfect equilibrium. In 2018, for example, two research teams independently made a fascinating discovery. A gene of viral origin encodes for a protein that plays a key role in long-term memory formation by moving information between cells in the nervous system. 9
P. Forterre (2008): Historically, three hypotheses have been proposed to explain the origin of viruses:
(1) they originated in a precellular world (‘the virus-first hypothesis’);
(2) they originated by reductive evolution from parasitic cells (‘the reduction hypothesis’); and
(3) they originated from fragments of cellular genetic material that escaped from cell control (‘the escape hypothesis’).
All these hypotheses had specific drawbacks. The virus-first hypothesis was usually rejected firsthand since all known viruses require a cellular host. The reduction hypothesis was difficult to reconcile with the observation that the most reduced cellular parasites in the three domains of life, such as Mycoplasma in Bacteria, Microsporidia in Eukarya, or Nanoarchaea in Archaea, do not look like intermediate forms between viruses and cells. Finally, the escape hypothesis failed to explain how such elaborate structures as complex capsids and nucleic acid injection mechanisms evolved from cellular structures since we do not know any cellular homologs of these crucial viral components.
Much like the concept of prokaryotes became the paradigm on how to think about bacterial evolution, the escape hypothesis became the paradigm favored by most virologists to solve the problem of virus origin. This scenario was chosen mainly because it was apparently supported by the observation that modern viruses can pick up genes from their hosts. In its classical version, the escape theory suggested that bacteriophages originated from bacterial genomes and eukaryotic viruses from eukaryotic genomes. This led to a damaging division of the virologist community into those studying bacteriophages and those studying eukaryotic viruses, ‘phages’ and viruses being somehow considered to be completely different entities. The artificial division of the viral world between ‘viruses’ and bacteriophages also led to much confusion on the nature of archaeal viruses. Indeed, although most of them are completely unrelated to bacterial viruses, they are often called ‘bacteriophages’, since archaea (formerly archaebacteria) are still considered by some biologists as ‘strange bacteria’. For instance, archaeal viruses are grouped with bacteriophages in the drawing that illustrates viral diversity in the last edition of the Virus Taxonomy Handbook. Hopefully, these outdated visions will finally succumb to the accumulating evidence from molecular analyses.
Viruses Are Not Derived from Modern Cells Abundant data are now already available to discredit the escape hypothesis in its classical adaptation of the prokaryote/eukaryote paradigm. This hypothesis indeed predicts that proteins encoded by bacterial viruses (avoiding the term bacteriophage here) should be evolutionarily related to bacterial proteins, whereas proteins encoded by viruses infecting eukaryotes should be related to eukaryotic proteins. This turned out to be wrong since, with a few exceptions (that can be identified as recent transfers from their hosts), most viral encoded proteins have either no homologs in any cell or only distantly related homologs. In the latter cases, the most closely related cellular homolog is rarely from the host and can even be from cells of a domain different from the host. More and more biologists are thus now fully aware that viruses form a world of their own, and that it is futile to speculate on their origin in the framework of the old prokaryote/ eukaryote dichotomy.
A more elaborate version has been proposed by William Martin and Eugene Koonin, who suggested that life originated and evolved in the cell-like mineral compartments of a warm hydrothermal chimney. In that model, viruses emerged from the assemblage of self-replicating elements using these inorganic compartments as the first hosts. The formation of true cells occurred twice independently only at the end of the process (and at the top of the chimney), producing the first archaea and bacteria. The latter escaped from the same chimney system as already fully elaborated modern cells. In the model, viruses first co-evolved with acellular machineries producing nucleotide precursors and proteins.
The emergence of the RNA world involves at least the existence of complex mechanisms to produce ATP, RNA, and proteins. This means an elaborated metabolism to produce ribonucleotide triphosphate (rNTP) and amino acids, RNA polymerases, and ribosomes, as well as an ATP-generating system. If such a complex metabolism was present, it appears unlikely that it was unable to produce lipid precursors, hence membranes. If this is correct, then ‘modern’ viruses did not predate cells but originated in a world populated by primitive cells.
Viruses and the Origin of DNA Considering the possibility that at least some DNA viruses originated from RNA viruses, it has been suggested that DNA itself could have appeared in the course of virus evolution (in the context of competition between viruses and their cellular hosts). Indeed, DNA is a modified form of RNA, and both viruses and cells often chemically modify their genomes to protect themselves from nucleases produced by their competitor. It is
usually considered that DNA replaced RNA in the course of evolution simply because it is more stable (thanks to the removal of the reactive oxygen in position 20 of the ribose) and because cytosine deamination (producing uracil) can be corrected in DNA (where uracil is recognized as an alien base) but not in RNA. 10
Anyone that studies biochemistry, knows the enormous complexity of ribonucleotide reductase enzymes, that remove oxygen from the 2' position of ribose, the backbone of RNA, to transform RNA into DNA. There is no scientific explanation for how RNA could have transitioned to DNA, and the origin of the ultra-complex machinery to catalyze the needed reactions. Molecules have no goals, no foresight. They did not think about the advantage of stability if transitioning to DNA. There's nothing about inert chemicals and physical forces that say we want to become part of a living self-replicating entity called a cell at the end of a chemical evolutionary process. Molecules do not have the "drive", they do not urge or "want" to find ways to become information-bearing biomolecules, or able to harness energy as ATP molecules, become more efficient, or become part of a molecular machine, or in the end, a complex organism. There is a further hurdle to overcome. More and more biologists are now fully aware that viruses form a world of their own. Proteins encoded by bacterial viruses are not related to bacterial proteins. Modern viruses exhibit very different types of genomes (RNA, DNA, single-stranded, double-stranded), including highly modified DNA, whereas all modern cellular organisms have double-stranded DNA genomes. So the question becomes how Viruses that have a DNA genome originated since they had an independent origin from living cells. Even more: P. Forterre (2008): Many DNA viruses encode their own enzymes for deoxynucleotide triphosphate (dNTP) production, ribonucleotide reductases (the enzymes that produce deoxyribonucleotides from ribonucleotides), and thymidylate synthases (the enzymes that produce deoxythymidine monophosphate (dTMP) from deoxyuridine monophosphate (dUMP).
That means RNR enzymes would have evolved independently, in a convergent manner,
twice !! Forterre continues: The replacement of RNA by DNA as cellular genetic material would have thus allowed genome size to increase, with a concomitant increase in cellular complexity (and efficiency) leading to the complete elimination of RNA cells by the ancestors of modern DNA cells. This traditional textbook explanation has been recently criticized as incompatible with Darwinian evolution since it does not explain what immediate selective advantage allowed the first organism with a DNA genome to predominate over former organisms with RNA genomes. Indeed, the newly emerging DNA cell could not have immediately enlarged its genome and could not have benefited straight away from a DNA repair mechanism to remove uracil from DNA. Instead, if the replacement of RNA by DNA occurred in the framework of the competition between cells and viruses, either in an RNA virus or in an RNA cell, modification of the RNA genome into a DNA genome would have immediately produced a benefit for the virus or the cell. It has been argued that the transformation of RNA genomes into DNA genomes occurred preferentially in viruses because it was simpler to change in one step the chemical composition of the viral genome than that of the cellular genomes (the latter interacting with many more proteins). Furthermore, modern viruses exhibit very different types of genomes (RNA, DNA, single-stranded, double-stranded), including highly modified DNA, whereas all modern cellular organisms have double-stranded DNA genomes. This suggests a higher degree of plasticity for viral genomes compared to cellular ones. The idea that DNA originated first in viruses could also explain why
many DNA viruses encode their own enzymes for deoxynucleotide triphosphate (dNTP) production, ribonucleotide reductases (the enzymes that produce deoxyribonucleotides from ribonucleotides), and thymidylate synthases (the enzymes that produce deoxythymidine monophosphate (dTMP) from deoxyuridine monophosphate (dUMP). Because in modern cells, dTMP is produced from dUMP, the transition from RNA to DNA occurred likely in two steps, first with the appearance of ribonucleotide reductase and production of U-DNA (DNA containing uracil), followed by the appearance of thymidylate synthases and formation of T-DNA (DNA containing thymine). The existence of a few bacterial viruses with U-DNA genomes has been taken as evidence that they could be relics of this period of evolution. If DNA first appeared in the ancestral virosphere, one has also to explain how it was later on transferred to cells. One scenario posits the co-existence for some time of an RNA cellular chromosome and a DNA viral genome (episome) in the same cell, with the progressive transfer of the information originally carried by the RNA chromosome to the DNA ‘plasmid’ via retro-transposition. 10
Viruses, the most abundant biological entities on earthSteven W. Wilhelm (2012): Viruses are the most abundant life forms on Earth, with an estimated 10^31 total viruses globally. 11 Eugene V. Koonin (2020): Viruses appear to be the dominant biological entities on our planet, with the total count of virus particles in aquatic environments alone at any given point in time reaching the staggering value of 10^31, a number that is at least an order of magnitude greater than the corresponding count of cells. The genetic diversity of viruses is harder to assess, but, beyond doubt, the gene pool of viruses is, in the least, comparable to that of hosts. The estimates of the number of distinct prokaryotes on earth differ widely, in the range of 10^7 to 10^12, and accordingly, estimation of the number of distinct viruses infecting prokaryotes at 10^8 to 10^13 is reasonable. Even assuming the lowest number in this range and even without attempting to count viruses of eukaryotes, these estimates represent vast diversity. Despite the rapid short-term evolution of viruses, the key genes responsible for virion formation and virus genome replication are conserved over the long term due to selective constraints. Genetic parasites inescapably emerge even in the simplest molecular replicator systems and persist through their subsequent evolution. Together with the ubiquity and enormous diversity of viruses in the extant biosphere, these findings lead to the conclusion that viruses and other mobile genetic elements MGEs played major roles in the evolution of life ever since its earliest stages.7 G.Witzany (2015): If we imagine that 1ml of seawater contains one million bacteria and ten times more viral sequences it can be determined that 10^31 bacteriophages infect 10^24 bacteria per second. 12
No common ancestor for VirusesEugene V. Koonin (2020): In the genetic space of viruses and mobile genetic elements (MGEs), no genes are universal or even conserved in the majority of viruses. Viruses have
several distinct points of origin, so there has never been a last common ancestor of all viruses. 7
Viruses and the tree of life ( Virology blog 2009): Viruses are polyphyletic (a group whose members come from multiple ancestral sources): In a phylogenetic tree, the characteristics of members of taxa are inherited from previous ancestors. Viruses cannot be included in the tree of life because they
do not share characteristics with cells, and no single gene is shared by all viruses or viral lineages. Viruses are polyphyletic – they have many evolutionary origins. Viruses don’t have a structure derived from a common ancestor. Cells obtain membranes from other cells during cell division. According to this concept of ‘membrane heredity’, today’s cells have inherited membranes from the first cells. Viruses have no such inherited structure. They play an important role in regulating population and biodiversity. 13
Eugene V. Koonin (2017):
The entire history of life is the story of virus-host coevolution. Therefore the origins and evolution of viruses are an essential component of this process. A signature feature of the virus state is the capsid, the proteinaceous shell that encases the viral genome. Although homologous capsid proteins are encoded by highly diverse viruses,
there are at least 20 unrelated varieties of these proteins. Viruses are the most abundant biological entities on earth and show a remarkable diversity of genome sequences, replication and expression strategies, and virion structures. Virus genomes typically consist of distinct structural and replication modules that recombine frequently and can have different evolutionary trajectories. 14
The importance of the admission that viruses do not share a common ancestor cannot be outlined enough. Researchers also admit, that under a naturalistic framework, the origin of viruses remains obscure, and has not found an explanation. One reason is that viruses depend on a cell host in order to replicate. Another is, that the virus capsid shells that protect the viral genome are unique, there is no counterpart in life. A science paper that I quote below describes capsids with a "geometrically sophisticated architecture not seen in other biological assemblies". This seems to be interesting evidence of design. The claim that their origin has something to do with evolution is also misleading - evolution plays no role in explaining either the origin of life or the origin of viruses. The fact that "no single gene is shared by all viruses or viral lineages" prohibits drawing a tree of viruses leading to a common ancestor.
Edward C. Holmes (2011): The discovery of mimivirus has undoubtedly had a major impact on theories of viral origins. More striking is that most (∼70% at the time of writing) mimivirus genes have no known homologs, in either virus or cellular genomes, so
their origins are unknown. More importantly, the discovery of mimivirus highlights our profound ignorance of the virosphere. It is therefore a truism that a wider sampling of viruses in nature is likely to tell us a great deal more about viral origins. Although perhaps less lauded, the discovery of conserved protein structures among diverse viruses with little if any primary sequence similarity has even grander implications for our understanding of viral origins. 15
Capsid-encoding organisms in contrast to ribosome-encoding organismsEugene V. Koonin (2014): Viruses were defined as one of the two principal types of organisms in the biosphere, namely, as
capsid-encoding organisms in contrast to r
ibosome-encoding organisms, i.e., all cellular life forms. Structurally similar, apparently homologous capsids are present in a huge variety of icosahedral viruses that infect bacteria, archaea, and eukaryotes. These findings prompted the concept of the capsid as the virus “self” that defines the identity of deep, ancient viral lineages. This “capsidocentric” perspective on the virus world is buttressed by observations on the extremely wide spread of certain capsid protein (CP) structures that are shared by an enormous variety of viruses, from the smallest to the largest ones, that infect bacteria, archaea, and all divisions of eukaryotes. The foremost among such conserved capsid protein structures is the so-called jelly roll capsid (JRC) protein fold, which is represented, in a variety of modifications, in extremely diverse icosahedral (spherical) viruses that infect hosts from all major groups of cellular life forms. In particular, the presence of the double-beta-barrel JRC (JRC2b) in a broad variety of double-stranded DNA (dsDNA) viruses infecting bacteria, archaea, and eukaryotes has been touted as an argument for the existence of an “ancient virus lineage,” of which this type of capsid protein is the principal signature. Under this approach, viruses that possess a single beta-barrel JRC (JRC1b)—primarily RNA viruses and single-stranded DNA (ssDNA) viruses— could be considered another major viral lineage. A third lineage is represented by dsDNA viruses with icosahedral capsids formed by the so-called HK97-like capsid protein (after bacteriophage HK97, in which this structure was first determined), with a fold that is unrelated to the jelly roll fold. This assemblage of viruses is much less expansive than those defined by either JRC2b or JRC1b, but nevertheless, it unites dsDNA viruses from all three domains of cellular life. The capsid-based definition of a virus does capture a quintessential distinction between the two major empires of life forms, i.e., viruses and cellular life forms. 16
Viruses with a different genetic alphabetStephen Freeland (2022): The genetic material of more than 200 bacteriophage viruses uses 1-aminoadenine (Z) instead of adenine (A). This minor difference in chemical structures is nevertheless a
fundamental deviation from the standard alphabet of four nucleobases established by biological evolution at the time of life's Last Universal Common Ancestor (LUCA). Placed into broader context, the finding illustrates a
deep shift taking place in our understanding of the chemical basis for biology. 17
What is the best explanation for viral origin?Edward C. Holmes (2011): The central debating point in discussions of the origin of viruses is whether they are ancient, first appearing before the last universal cellular ancestor (LUCA), or evolved more recently, such that their ancestry lies with genes that “escaped” from the genomes of their cellular host organisms and subsequently evolved independent replication. The escaped gene theory has traditionally dominated thinking on viral origins, in large part because viruses are parasitic on cells now and it has been argued that this must have always have been the case. However,
there is no gene shared by all viruses, and recent data are providing increasingly strong support for a far more ancient origin. 18
Koonin mentions three possible scenarios for their origin. One of them: Eugene V. Koonin (2017):
The virus-first hypothesis, also known as the primordial virus world hypothesis, regards viruses (or virus-like genetic elements) as intermediates between prebiotic chemical systems and cellular life and accordingly posits that virus-like entities originated in the precellular world.The second:
The regression hypothesis, in contrast, submits that viruses are degenerated cells that have succumbed to obligate intracellular parasitism and in the process shed many functional systems that are ubiquitous and essential in cellular life forms, in particular the translation apparatus. The third, the escape hypothesis postulates that viruses evolved independently in different domains of life from cellular genes that embraced selfish replication and became infectious. 14
The second and third are questionable, in face of the fact that evolution would sort out degenerated cell parts that would harm their survival. The hypothesis that these parts would become parasites, goes detrimentally against the evolutionary paradigm, since evolution is about the survival of the fittest, and not evolving parasites that would kill the cell. Furthermore, if Viruses were not extant right from the beginning, how would ecological homeostasis be guaranteed?
Koonin agrees that the first is the most plausible. He writes: The diversity of genome replication-expression strategies in viruses, contrasting the uniformity in cellular organisms, had been considered to be most compatible with the possibility that the virus world descends directly from a precellular stage of evolution, and an updated version of the escape hypothesis states that the first viruses have escaped not from contemporary but rather from primordial cells, predating the last universal cellular ancestor. The three evolutionary scenarios imply different timelines for the origin of viruses but offer little insight into how the different components constituting viral genomes might have combined to give rise to modern viruses.
The conclusion that can be drawn is, that Viruses co-emerged with life, and that occurred multiple times. If just emerging once is extremely unlikely based on the odds, how much more, multiple times?
Koonin continues: A typical virus genome encompasses two major functional modules, namely,
determinants of virion formation and those of
genome replication. Understanding the origin of any virus group is possible only if the provenances of both components are elucidated. Given that viral replication proteins often have no closely related homologs in known cellular organisms, it has been suggested that many of these proteins evolved in the precellular world or in primordial, now extinct, cellular lineages. The ability to transfer the genetic information encased within capsids—the protective proteinaceous shells that comprise the cores of virus particles (virions)—is unique to bona fide viruses and distinguishes them from other types of selfish genetic elements such as plasmids and transposons. Thus, the origin of the first true viruses is inseparable from the emergence of viral capsids. Studies on the origin of viral capsids are severely hampered by the high sequence divergence among these proteins.
Analysis of the available sequences and structures of major capsid proteins (CP) and nucleocapsid (NC) proteins encoded by representative members of 135 virus taxa (117 families and 18 unassigned genera) allowed us to attribute structural folds to 76.3% of the known virus families and unassigned genera. The remaining taxa included viruses that do not form viral particles (3%) and viruses for which the fold of the major virion proteins is not known and could not be predicted from the sequence data (20.7%). The former group includes capsidless viruses of the families
Endornaviridae, Hypoviridae, Narnaviridae, and Amalgaviridae, all of which appear to
have evolved independently from different groups of full-fledged capsid-encoding RNA viruses. The latter category includes eight taxa of archaeal viruses with
unique morphologies and genomes, pleomorphic bacterial viruses of the family Plasmaviridae, and 19 diverse taxa of eukaryotic viruses. It should be noted that, with the current explosion of metagenomics studies, the number and diversity of newly recognized virus taxa will continue to rise. Although many of these viruses are expected to have previously observed CP/NC protein folds, novel architectural solutions doubtlessly will be discovered as well. 17
Gladys Kostyrka (2016): To french molecular biologist and microbiologist Patrick Forterre,
viruses could not exist without cells because he endorses their definition as intracellular obligate parasites. However, this does not mean that viruses did not exist prior to DNA cells. On the basis of comparative sequence analyses of proteins and nucleic acids from viruses and their cellular hosts, Forterre hypothesized that viruses originated before DNA cells and before LUCA (the Last Universal Cellular Ancestor). Forterre’s hypothesis has been first formulated in the 1990s and was inspired by protein phylogenies. “Comparative sequence analyses of type II DNA topoisomerases and DNA polymerases from viruses, prokaryotes and eukaryotes suggest that viral genes diverged from cellular genes before the emergence of the last common ancestor (LCA) of prokaryotes and eukaryotes”. At least some viruses originated not from the known cellular domains e Bacteria, Eukarya, and Archaea e but before these three domains were formed. In other words, these viruses must have originated before LUCA. There are several genes shared by many groups of viruses with extremely diverse replication-expression strategies, genome size and host ranges. In other words, there are several “hallmark genes”, coding for several hallmark proteins present in many viruses. Yet these genes and proteins are not supposed to be shared by viruses that do not have the same origin, given their diversity. This “key observation” of several hallmark viral genes is thus problematic. It is even more problematic if one takes into account the fact that these genes are not found in any cellular life forms. It is then highly improbable that these viral hallmark genes were originally cellular genes that were transferred to viruses. Koonin assumes that these genes originated in a primordial viral world and were conserved. “The simplest explanation for the fact that the hallmark proteins involved in viral replication and virion formation are present in a broad variety of viruses but not in any cellular life forms seems to be that the latter actually never possessed these genes. Rather, the hallmark genes, probably, antedate cells and descend directly from the primordial pool of virus-like genetic elements” 19
If Koonin's hypothesis were the case, these nucleotides would require foresight to assemble into genes, that later would become virions, depending on cell hosts. That seems not tenable. The evidence is better interpreted by the creationist model. It coincides with the hypothesis, that God created each species/kind and virus separately. Multiple creation events by natural means and the emergence of symbiotic and parasitic relationships just mean multiplying the odds, and then naturalistic proposals become more and more untenable.
Achieving the same function through different molecular assembly routes refutes an evolutionary-naturalistic origin of virusesEugene V. Koonin (2015): The ability to form virions is the key feature that distinguishes viruses from other types of mobile genetic elements, such as plasmids and transposons. The origin of bona fide viruses thus appears to be intimately linked to and likely concomitant with the origin of the capsids. However, tracing the provenance of viral capsid proteins (CPs) proved to be particularly challenging because they typically do not display sequence or structural similarity to proteins from cellular life forms. Over the years, a number of structural folds have been discovered in viral CPs. Strikingly, morphologically similar viral capsids, in particular, icosahedral, spindle-shaped and filamentous ones, can be built from CPs which have unrelated folds. Thus, viruses have found multiple solutions to the same problem. Nevertheless, the process of de novo origin of viral CPs remains largely enigmatic. 9
Stephen J. Gould (1990):…No finale can be specified at the start, none would ever occur a second time in the same way, because any pathway proceeds through thousands of improbable stages. Alter any early event, ever so slightly, and without apparent importance at the time, and evolution cascades into a radically different channel.21
Fazale Rana (2001): Gould’s metaphor of “replaying life’s tape” asserts that if one were to push the rewind button, erase life’s history, and let the tape run again, the results would be completely different. The very essence of the evolutionary process renders evolutionary outcomes as nonreproducible (or nonrepeatable). Therefore, “repeatable” evolution is inconsistent with the mechanism available to bring about biological change. 22
William Schopf (2002): Because biochemical systems comprise many intricately interlinked pieces, any particular full-blown system can only arise once…Since any complete biochemical system is far too elaborate to have evolved more than once in the history of life, it is safe to assume that microbes of the primal LCA cell line had the same traits that characterize all its present-day descendants. 23 24
Hugh M. B. Harris: (2021): Viruses are ubiquitous. They infect almost every species and are probably the most abundant biological entities on the planet, yet they are excluded from the Tree of Life (ToL).
Viruses may well be essential for ecosystem diversity 25
Matti Jalasvuori (2012): Viruses play a vital role in all cellular and genetic functions, and
we can therefore define viruses as essential agents of life. Viruses provide the largest reservoir of genes known in the biosphere but were not, stolen’ from the host. Such capsids cannot be of host origin. It is well accepted by virologists that viruses often contain many complex genes (including core genes) that cannot be attributed to having been derived from host genes. 26
Julia Durzyńska (2015): Many attempts have been made to define nature of viruses and to uncover their origin. As the origin of viruses and that of living cells are
most probably interdependent, we decided to reveal ideas concerning nature of cellular last universal common ancestor (LUCA). Many viral particles (virions) contain specific viral enzymes required for replication. A few years ago, a new division for all living organisms into two distinct groups has been proposed:
ribosome-encoding organisms (REOs) and capsid-encoding organisms (CEOs). 27
Eugene V. Koonin: (2012): Probably an even more fundamental departure from the three-domain schema is the discovery of the Virus World, with its unanticipated, astonishing expanse and equally surprising evolutionary connectedness. Virus-like parasites inevitably emerge in any replicator systems, so
THERE IS NO EXAGGERATION IN THE STATEMENT THAT THERE IS NO LIFE WITHOUT VIRUSES. And in quite a meaningful sense, not only viruses taken together, but also major groups of viruses seem to be no less (if not more) fundamentally distinct as the three (or two) domains of cellular life forms, given that viruses employ different replication-expression cycles, unlike cellular life forms which, in this respect, are all the same. 28
Shanshan Cheng: (2013): Viral capsid proteins protect the viral genome by forming a closed protein shell around it. Most of currently found viral shells with known structure are spherical in shape and observe icosahedral symmetry. Comprised of a large number of proteins, such large, symmetrical complexes assume a
geometrically sophisticated architecture not seen in other biological assemblies. Geometry of the complex architecture aside, another striking feature of viral capsid proteins lies in the folded topology of the monomers, with the canonical jelly-roll β barrel appearing most prevalent (but not sole) as a core structural motif among capsid proteins that make up these viral shells of varying sizes. Our study provided support for the hypothesis that viral capsid proteins, which are
functionally unique in viruses in constructing protein shells, are also structurally unique in terms of their folding topology.29Eugene V. Koonin (2020): In a seminal 1971 article, Baltimore classified all then-known viruses into six distinct classes that became known as Baltimore classes (BCs) (a seventh class was introduced later), on the basis of the structure of the virion's nucleic acid (traditionally called the virus genome):
The seven Baltimore classes (BCs)For each BC, the processes of replication, transcription, translation, and virion assembly are shown by color-coded arrows (see the inset). Host enzymes that are involved in virus genome replication or transcription are prefixed with “h-,” and in cases when, in a given BC, one of these processes can be mediated by either a host- or a virus-encoded enzyme, the latter is prefixed with “v-.” Otherwise, virus-encoded enzymes are not prefixed. CP, capsid protein; DdDp, DNA-directed DNA polymerase; DdRp, DNA-directed RNA polymerase; gRNA, genomic RNA; RdRp, RNA-directed RNA polymerase; RT, reverse transcriptase; RCRE, rolling-circle replication (initiation) endonuclease.
1. Double-stranded DNA (dsDNA) viruses, with the same replication-expression strategy as in cellular life forms
2. Single-stranded DNA (ssDNA) viruses that replicate mostly via a rolling-circle mechanism
3. dsRNA viruses
4. Positive-sense RNA [(+)RNA] viruses that have ssRNA genomes with the same polarity as the virus mRNA(s)
5. Negative-sense RNA [(−)RNA] viruses that have ssRNA genomes complementary to the virus mRNA(s)
6. RNA reverse-transcribing viruses that have (+)RNA genomes that replicate via DNA intermediates synthesized by reverse transcription of the genome
7. DNA reverse-transcribing viruses replicating via reverse transcription but incorporating into virions a dsDNA or an RNA-DNA form of the virus genome.
Evidence supports monophyly for some of the BCs but refutes it for others. Generally, the evolution of viruses and MGEs is studied with methods of molecular evolutionary analysis that are also used for cellular organisms. However, the organizations of the genetic spaces dramatically differ between viruses and their cellular hosts.
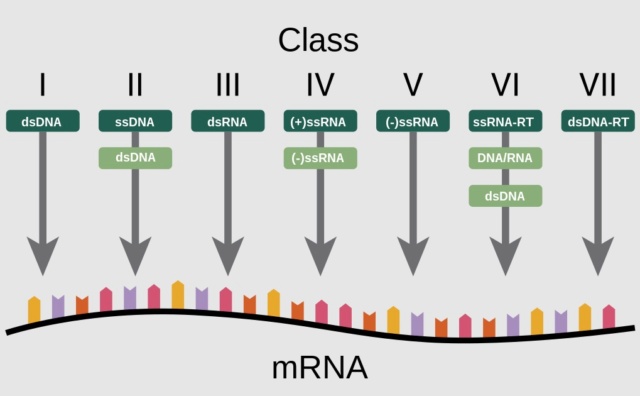
An illustration of the "pathways" each Baltimore group goes through to synthesize mRNA. Of the 6 “superviral hallmark genes” in virus genomes of the seven Baltimore classes.
Koonin (2020):The origins of superviral hallmark genes VHGs appear to be widely different. In particular, RdRps, RTs, and RCREs most likely represent the heritage of the primordial replicator pool as indicated by the absence of orthologs of these proteins in cellular life-forms.
At the top of the megataxonomy are the four effectively independent realms that, however, are connected at an even higher rank of unification through the super-VHG domains. 7
Rob Phillips (2018):The International Committee on Taxonomy of Viruses or ICTV classifies viruses into seven orders:
Herpesvirales, large eukaryotic double-stranded DNA viruses;
Caudovirales, tailed double-stranded DNA viruses typically infecting bacteria;
Ligamenvirales, linear double-stranded viruses infecting archaea;
Mononegavirales, nonsegmented negative (or antisense) strand single-stranded RNA viruses of plants and animals;
Nidovirales, positive (or sense) strand single-stranded RNA viruses of vertebrates;
Picornavirales, small positive strand single-stranded RNA viruses infecting plants, insects, and animals;
Tymovirales, monopartite positive single-stranded RNA viruses of plants.
In addition to these orders, there are ICTV families, some of which have not been assigned to an ICTV order. Only those ICTV viral families with more than a few members present in our dataset are explored. 30
Structure and Assembly of Complex VirusesCarmen San Martin (2013): Viral particles consist essentially of a proteinaceous capsid protecting a genome and involved also in many functions during the virus life cycle. In simple viruses, the capsid consists of a number of copies of the same, or a few different proteins organized into a symmetric oligomer. Structurally complex viruses present a larger variety of components in their capsids than simple viruses. They may contain accessory proteins with specific architectural or functional roles; or incorporate non-proteic elements such as lipids. They present a range of geometrical variability, from slight deviations from the icosahedral symmetry to complete asymmetry or even pleomorphism. Putting together the many different elements in the virion requires an extra effort to achieve correct assembly, and thus complex viruses require sophisticated mechanisms to regulate morphogenesis. This chapter provides a general view of the structure and assembly of complex viruses.
A viral particle consists essentially of a proteinaceous capsid with multiple roles in the protection of the viral genome, cell recognition and entry, intracellular trafficking, and controlled uncoating. Viruses adopt different strategies to achieve these goals. Simple viruses generally build their capsids from a number of copies of the same, or a few different proteins, organized into a symmetric oligomer. In the case of complex viruses, capsid assembly requires further elaborations. What are the main characteristics that define a structurally complex virus? Structural complexity on a virus often, but not necessarily, derives from the need to house a large genome, in which case a larger capsid is required. However, capsid or genome sizes by themselves are not determinants of complexity. For example, flexible filamentous viruses can reach lengths in the order of microns, but most of their capsid mass is built by a single capsid protein arranged in a helical pattern. On the other hand, architecturally complex viruses such as HIV have moderate-sized genomes (7–10 kb of single-stranded (ss) RNA). Structurally complex viruses incorporate a larger variety of components into their capsids than simple viruses. They may contain accessory proteins with specific architectural or functional roles or incorporate non-proteic elements such as lipids. 31
Forming viral symmetric shellsRoya Zandi (2020): The process of formation of virus particles in which the protein subunits encapsidate genome (RNA or DNA) to form a stable, protective shell called the capsid is an essential step in the viral life cycle. The capsid proteins of many small single-stranded RNA viruses spontaneously package their wild-type (wt) and other negatively charged polyelectrolytes, a process basically driven by the electrostatic interaction between positively charged protein subunits and negatively charged cargo. Regardless of the virion size and assembly procedures, most spherical viruses adopt structures with icosahedral symmetry. How exactly capsid proteins (CPs) assemble to assume a specific size and symmetry have been investigated for over half a century now. As the self-assembly of virus particles involves a wide range of thermodynamics parameters, different time scales, and an extraordinary number of possible pathways, the kinetics of assembly has remained elusive, linked to Levinthal’s paradox for protein folding. The role of the genome on the assembly pathways and the structure of the capsid is even more intriguing. The kinetics of virus growth in the presence of RNA is at least 3 orders of magnitude faster than that of empty capsid assembly, indicating that the mechanism of assembly of CPs around RNA might be quite different. Some questions then naturally arise: What is the role of RNA in the assembly process, and by what means then does RNA preserve assembly accuracy at fast assembly speed? Two different mechanisms for the role of the genome have been proposed: (i) en masse assembly and (ii) nucleation and growth.
The assembly interfaces in many CPs are principally short-ranged hydrophobic in character, whereas there is a strong electrostatic, nonspecific long-ranged interaction between RNA and CPs. To this end, the positively charged domains of CPs associate with the negatively charged RNA quite fast and form an amorphous complex. Hydrophobic interfaces then start to associate, which leads to the assembly of a perfect icosahedral shell. Based on the en masse mechanism, the assembly pathways correspond to situations in which intermediates are predominantly disordered. They found that, at neutral pH, a considerable number of CPs were rapidly (∼28 ms) adsorbed to the genome, which more slowly (∼48 s) self-organized into compact but amorphous nucleoprotein complexes (NPC). By lowering the pH, they observed a disorder−order transition as the protein−protein interaction became strong enough to close up the capsid and to overcome the high energy barrier separating NPCs from virions. 32
1. Curtis A. Suttle: Viruses in the sea 2005
2. Gladys Kostyrka: What roles for viruses in origin of life scenarios? 27 February 2016
3. Hugh M. B. Harris A Place for Viruses on the Tree of Life 14 January 2021
4. CARRIE ARNOLD: Could Giant Viruses Be the Origin of Life on Earth? JULY 17, 2014
5. Hugh Ross: Viruses and God’s Good Designs March 30, 2020
6. Ramesh K Goel: Viruses and Their Interactions With Bacteria and Archaea of Hypersaline Great Salt Lake 2021 Sep 28
7. Eugene V. Koonin: Global Organization and Proposed Megataxonomy of the Virus World 4 March 2020
8. Eugene VKoonin: A virocentric perspective on the evolution of life October 2013
9. Rachel Nuwer Why the world needs viruses to function (2020)
10. P.Forterre: Origin of Viruses 2008
11. Steven W. Wilhelm: Ocean viruses and their effects on microbial communities and biogeochemical cycles 2012 Sep 5.
12. G.Witzany: Viruses are essential agents within the roots and stem of the tree of life 21 February 2010
13. Viruses and the tree of life 19 March 2009
14. Eugene V. Koonin: Multiple origins of viral capsid proteins from cellular ancestors March 6, 2017
15. Edward C. Holmes: What Does Virus Evolution Tell Us about Virus Origins? 2011 Jun; 85
16. Eugene V. Koonin: Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements 2, June 2014
17. Stephen Freeland: Undefining life's biochemistry: implications for abiogenesis 23 February 2022
18. Edward C. Holmes: What Does Virus Evolution Tell Us about Virus Origins? 2011 Jun; 8519. Gladys Kostyrka: What roles for viruses in origin of life scenarios? 27 February 201620. Eugene V. Koonin: Evolution of an archaeal virus nucleocapsid protein from the CRISPR-associated Cas4 nuclease 2015
21. Stephen J. Gould, Wonderful Life: The Burgess Shale and the Nature of History 1990
22. Fazale Rana: Repeatable Evolution or Repeated Creation? 2001
23. J. William Schopf: Life’s Origin 2002
24. Fazale Rana: Newly Discovered Example of Convergence Challenges Biological Evolution 2008
25. Hugh M. B. Harris: A Place for Viruses on the Tree of Life 14 January 2021
26. Matti Jalasvuori Viruses: Essential Agents of Life (2012)
27. Julia Durzyńska Viruses and cells intertwined since the dawn of evolution (2015)
28. Eugene V. Koonin: The Logic of Chance : The Nature and Origin of Biological Evolution (2012)
29. Shanshan Cheng: Viral Capsid Proteins Are Segregated in Structural Fold Space February 7, 2013
30. Rob Phillips: A comprehensive and quantitative exploration of thousands of viral genomes 2018 Apr 19
31. Carmen San Martin: Structure and Assembly of Complex Viruses 19 April 2013
32. Roya Zandi: How a Virus Circumvents Energy Barriers to Form Symmetric Shells March 2, 2020

























