

The immune system, and irreducible complexity
https://reasonandscience.catsboard.com/t2322-the-immune-system-and-irreducible-complexity
Darwins black box, page 97
Although great strides have been made in understanding how the immune system works, we remain ignorant of how it came to be. None of the questions raised has been answered by any of the thousands of scientists in the field; few have even asked the questions. A search of the immunological literature shows ongoing work in comparative immunology (the study of immune systems from various species). But that work, valuable though it is, does not address in molecular detail the question of how immune systems originated.
Perhaps the best efforts at doing that so far have been in two short papers. The first, by Nobel laureate David Baltimore and two other prominent scientists, is tantalizingly entitled «Molecular Evolution of the Vertebrate Immune System.» But it's hard to live up to such a title in just two pages. The authors point out that for any organism to have an immune system akin to that seen in mammals, the minimally required molecules are the antigen receptors (immunoglobulin and TCR), the antigen presentation molecules (MHC), and the gene rearranging proteins. (Immunoglobulins are antibodies. TCR molecules are akin to antibodies.) The authors then argue that sharks, which are very distantly related to mammals, appear to have all three components.
But it's one thing to say an organism has a completed, functioning system, and another to say how the system developed. The authors certainly realize this. They note that immunoglobulin and TCR genes both require RAG proteins for rearrangement. On the other hand, RAG proteins require specific recombination signals to rearrange immunoglobulin and TCR genes. (RAG is the component that rearranges the genes.) They make a valiant stab at accounting for the components, but in the end, it is a hop in the box with Calvin and Hobbes. The authors speculate that a gene from a bacterium might have luckily been transferred to an animal. Luckily, the protein coded by the gene could itself rearrange genes; and luckily, in the animal's DNA there were signals that were near antibody genes; and so on. In the final analysis the authors identify key problems with gradualistic evolution of the immune system, but their proffered solutions are really just a disguised shrug of their shoulders.
I have looked at three features of the immune system—clonal selection, antibody diversity, and the complement system—and demonstrated that each individually poses massive challenges to a putative step-by-step evolution. But showing that the parts can't be built step by step only tells part of the story, because the parts interact with each other. Just as a car without steering, or a battery, or a carburetor isn't going to do you much good, an animal that has a clonal selection system won't get much benefit out of it if there is no way to generate antibody diversity. A large repertoire of antibodies won't do much good if there is no system to kill invaders. A system to kill invaders won't do much good if there's no way to identify them. At each step we are stopped not only by local system problems, but also by requirements of the integrated system.
THE RIGHT STUFF
When a microscopic invader breaches the outer defenses of the body the immune system swings into action. This happens automatically. The molecular systems of the body, like the Star Wars anti-missile system that the military once planned, are robots designed to run on autopilot. Since the defense is automated, every step has to be accounted for by some mechanism. The first problem that the automated defense system has is how to recognize an invader. Bacterial cells have to be distinguished from blood cells; viruses have to be distinguished from connective tissue. Unlike us, the immune system can't see, so it has to rely initially on something akin to a sense of touch. Antibodies are the «fingers» of the blind immune system—they allow it to distinguish a foreign invader from the body itself. Antibodies are formed by an aggregation of four chains of amino acids (Figure 6-1):
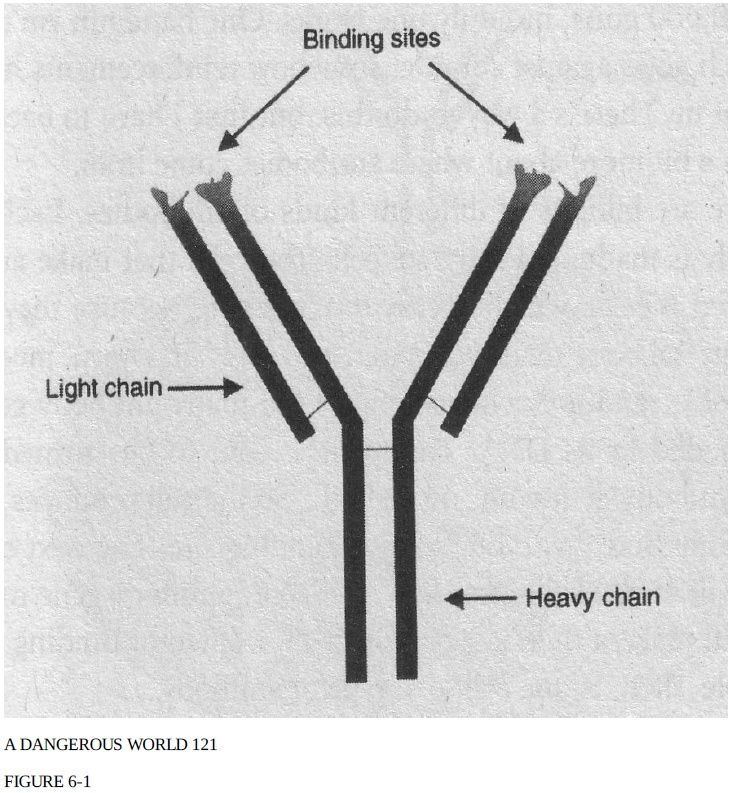
two identical light chains, and two identical heavy chains. The heavy chains are about twice as big as the light chains. In the cell, the four chains make a complex that resembles the letter Y. Because the two heavy chains are the same and the two light chains are the same, the Y is symmetrical: if you took a knife and cut it down the middle you'd get identical halves, with one heavy and one light chain in each half. At the end of each pronged tip of the Y there is a depression (called a binding site). Lining the binding site are portions of both the light chain and the heavy chain. Binding sites come in a large variety of shapes. One antibody might have a binding site with a piece jutting up here, a hole over there, and an oily patch on the edge. A second antibody might have a positive charge on the left, a crevice in the middle, and a bump on the right.
If the shape of a binding site just happens to be exactly complementary to the shape of a molecule on the surface of an invading virus or bacterium, then the antibody will bind to that molecule. To get a feel for it, imagine a household object with a depression in it and a few knobs poking up out of the depression. My youngest daughter has a doll wagon with front and back seats—something like that will do nicely. Now take the wagon/object, go around the house, and see how many other articles will fit snugly into the depression, filling both the front seat and the back seat without leaving any spaces. If you find even one, you're luckier than I am. Nothing in my house fit snugly in the wagon, and neither did anything in my office or laboratory. I imagine there's some object out in the world with a shape complementary to the wagon's, but I haven't found it yet. The body has a similar problem: the odds of any given antibody binding to any given invader are pretty slim. To make sure that at least one kind of antibody is available for each attacker, we make billions to trillions of them. Usually, for any particular invader; it takes 100,000 to find one antibody that works. When bacteria invade the body, they multiply.
By the time an antibody binds to a bacterium there may be many, many copies of the bug floating around. Against this Trojan horse that breeds, the body has 100,000 guns, but only one works. One handgun isn't going to do much good against a horde; somehow reinforcements have to be brought in. There's a way to do this, but first I have to back up and explain a bit more about where antibodies come from. There are billions of different kinds of antibodies. Each kind of antibody is made in a separate cell. The cells that make antibodies are called В cells, which is easy to remember because they are produced in the bone marrow. When а В cell is first born, mechanisms inside of it randomly choose one of the many antibody genes that are encoded in its DNA. That gene is said to be «turned on»; all other antibody genes are «turned off.» So the cell produces only one kind of antibody, with one kind of binding site. The next cell that's made will in all likelihood have a different antibody gene turned on, so it will make a different protein with a different binding site.
The principle, then, is one cell, one type of antibody. Once a cell commits to making its antibody, you might think that the antibody would leave the cell so it could patrol the body. But if the contents of all В cells were dumped out into the body, there would be no way to tell which cell the antibody came from. The cell is the factory that makes the particular type of antibody; if the antibody finds a bacterium, we need to tell the cell to send us reinforcements. But with this hypothetical setup, we can't get a message back. Fortunately, the body is smarter than that. When а В cell first makes its antibody, the antibody anchors in the cell membrane with the prongs of the Y sticking out (Figure 6-2).

The cell does this trick by using the gene for the normal antibody, and also using a little piece of a gene that codes for an oily tail on the protein. Since the membrane is oily, too, the piece sticks in the membrane. This step is critical, because now the binding site of the antibody is attached to its factory. The entire В cell factory patrols the body; when a foreign invader enters, the antibody-with-attached-cell binds. Now we have the factory close at hand to the invaders. If the cell could be signaled to make more of the antibody, then the fight would be helped by reinforcements. Fortunately, there is a way to send a signal; unfortunately, it's pretty convoluted. When an antibody on а В cell binds to a foreign molecule it triggers a complex mechanism to swallow the invader: in effect, the munitions factory takes a hostage. The antibody then breaks off a piece of membrane to make a little vesicle—a self-made taxicab. In this taxi, the hostage is brought into the B-cell factory. Inside the cell (still in the cab) the foreign protein is chopped up, and a piece of the foreign protein sticks to another
protein (called an MHC protein). The cab then returns to the membrane of the cell. Outside the factory, along comes another cell (called a helper T cell). The helper T cell binds to the В cell, which is «presenting» the chopped-up piece of invader (the foreign fragment in the MHC protein) for the T cell's consideration. If the fit is just right, it causes the helper T cell to secrete a substance called interleukin. Interleukin is like a message from the Department of Defense to the munitions factory. By binding to another protein on the surface of the В cell, the interleukin sets off a chain of events that sends a message to the nucleus of the В cell. The message is: grow!
The В cell begins to reproduce at a rapid rate. T cells continue to secrete interleukin if they are bound to а В cell. Eventually the growing B-cell factory produces a series of spinoff factories in the form of specialized cells called 'plasma cells.' Instead of producing a form of the antibody that sticks in the membrane, plasma cells leave off the last oily piece of the protein. Now free antibody is extruded in large amounts into the extracellular fluid. The switch is critical. If the new plasma-cell factories were like the old B-cell factory, the antibodies would all be confined to quarters and would be much less effective at inhibiting the invaders.
STEP BY STEP
Could this system have evolved step-by-step? Consider the vast pool billions to trillions of factory В cells. The process of picking the right cell out of a mixture of antibody-producing cells is called clonal selection. Clonal selection is an elegant way to mount a specific response in great numbers to a wide variety of possible foreign invaders. The process depends on a large number of steps, some of which I have not discussed yet. Leaving those aside for now, let's ask what the minimum requirements are for a clonal selection system, and if those minimum requirements could be produced step-by-step. The key to the system is the physical connection of the binding ability of the protein with the genetic information for the protein. Theoretically this could be accomplished by making an antibody where the tail of the Y bound to the DNA that coded for the protein. In real life, however, such a setup wouldn't work. The protein might be connected to its genetic information, but because the cell is surrounded by a membrane, the antibody would never come in contact with the foreign material, which is floating around outside the cell. A system where both the antibody and its attached gene were exported from the cell would overcome that problem, only to run into a different one: outside the cell there would be no cellular machinery to translate the DNA message into more protein. Anchoring the antibody in the membrane is a good solution to the problem; now the antibody can mix it up with a foreign cell and still be near its DNA. But although the antibody can bind the foreign material without floating away from the cell, it does not have direct physical contact with the DNA. Since the protein and DNA are blind, there must be a way to get a message from one to the other. Just for now, for the sake of argument, let's forget about the tortuous way that the message of binding actually gets to the B-cell nucleus (requiring the taxicab, ingestion, MHC, helper T cells, interleukin, and so on).
Instead let's imagine a simpler system where there's only one other protein. Let's say that when the antibody binds to a foreign molecule, something happens that attracts some other protein—a messenger to take word of a hostage to the factory nucleus. Maybe when the hostage is first found, the shape of the antibody changes, perhaps pulling up a little on the antibody's tail. Perhaps part of the antibody's tail sticks into the inside of the cell, which is what triggers the messenger protein. The change in the tail could cause the messenger protein to scuttle into the nucleus and bind to the DNA at a particular point. Binding to the right place on the DNA is what causes the cell to start growing and to start producing antibody without the oily tail—antibody that gets sent out of the cell to fight the invasion. Even in such a simplified scheme, we are left with three critical ingredients:
(1) the membrane-bound form of the antibody;
(2) the messenger; and
(3) the exported form of the antibody.
If any of these components is missing, the system fails to function. If there is no antibody in the membrane, then there's no way to connect a successful antibody that binds a foreign invader to the cell containing the genetic information. If there is no exported form of the antibody, then when the signal is received there is nothing to send out into the world to fight. If there is no messenger protein, then there is no connection between binding the membrane antibody and turning on the right gene (making the system about as useful as a doorbell whose wires had been cut). A cell hopefully trying to evolve such a system in gradual Darwinian steps would be in a quandary. What should it do first? Secreting a little bit of antibody into the great outdoors is a waste of resources if there's no way to tell if it's doing any good. Ditto for making a membrane-bound antibody. And why make a messenger protein first if there is nobody to give it a message, and nobody to receive the message if it did get one? We are led inexorably to the conclusion that even this greatly simplified clonal selection could not have come about in gradual steps.
Even at this simplified level, then, all three ingredients had to evolve simultaneously. Each of these three items—the fixed antibody, the messenger protein, and the loose antibodies—had to be produced by a separate historical event, perhaps by a coordinated series of mutations changing preexisting proteins that were doing other chores into the components of the antibody system. Darwin's small steps have become a series of wildly unlikely leaps.
Yet our analysis overlooked many complexities: How does the cell switch from putting the extra oily piece on the membrane to not putting it on? The message system then is fantastically more complicated than our simplified version. Ingestion of the protein, chopping it up, presenting it to the outside on an MHC protein, specific recognition of the МНС/fragment by a helper T cell, secretion of interleukin, binding of interleukin to the В cell, sending the signal that interleukin has bound into the nucleus— the prospect of devising a step-by-step pathway for the origin of the system is enough to make strong men blanch.Factories float around in huge numbers, poised to deliver antibodies that can stick to an invader with virtually any shape. But how does the body make all those billions of differently shaped antibodies? It turns out that there is an elegant trick for making very many different antibodies without requiring enormous quantities of genetic material to code for the proteins.
Again, don't be concerned if the details quickly slip your mind; my purpose here is just to help you appreciate the complexity of the immune system. It took a fascinating discovery to lead scientists to puzzle out the full complexity of the immune system. The discovery started with a potentially cruel, but necessary, experiment. Just to see what would happen, chemists made some small molecules that do not occur in nature and then attached them to a protein. When the protein carrying the synthetic molecules was injected into a rabbit, the scientists were astonished to find that, yes, the rabbit made antibodies that bound tightly to the synthetic molecule. How could this be? Neither the rabbit nor its ancestors ever met the synthetic molecule, so how did it know how to make antibodies against it? Why should it recognize a molecule it had never seen before?
The puzzle of «antibody diversity» intrigued scientists studying immunology. Several ideas were floated as possible explanations. Proteins were known to be flexible molecules, and antibodies are proteins. So maybe when a new molecule is injected into the body an antibody wraps around it, molds itself to that shape, and then somehow freezes in that configuration. Or maybe, because defense is so vitally important, the DNA of organisms contains a vast number of genes for antibodies with many different shapes—enough to allow them to recognize things they hadn't seen yet. But such a huge number of antibodies would take up more than the available coding space in the DNA. So maybe there were only a few antibodies, and when the cell divided, maybe there was some way to make a lot of mutations in just the areas coding for the binding sites of the antibodies. That way each new В cell in the body could carry different mutations, coding for an antibody different from all other В cells. Or maybe the answer was a combination of these, or maybe it involved something completely new.
The answer to the problem of antibody diversity had to await an astonishing discovery: a gene coding for a protein didn't always have to be a continuous segment of DNA—it could be interrupted. If we compare a gene to a sentence, it was as if a protein's code, «The quick brown fox jumps over the lazy dog» could be altered (without destroying the protein) to read «The quick brdkdjf bufjwkw nhruown fox jumps over the lapfeqmzda lfybnek sybagjufu zy dog.» The sensible DNA message was broken up by tracts of nonsense letters that somehow were not included in the protein. Further work showed that for most genes, corrections would be made— splicing out the nonsense—after an RNA copy is made of a DNA gene.
Even with «interrupted» DNA, an edited and corrected message in RNA could be used by the cell's machinery to make the correct protein. Even more surprisingly, for antibody genes the DNA itself can also be spliced. In other words, DNA that is inherited can be altered. Amazing! Splicing and rearrangement of DNA play a large role in explaining the great number of antibodies that the body can produce.
The following is a brief description of work that has taken many investigators many years to accomplish; because of their efforts, the riddle of antibody diversity is solved.
At conception there are a number of gene pieces in the fertilized cell that contribute to making antibodies. The genes are arranged into clusters that I will simply call cluster 1, cluster 2, and so forth. In humans there are approximately 250 gene segments in cluster 1; a ways down the DNA from cluster 1 are ten gene segments that form cluster 2; further on down the DNA road are a group of six segments that comprise cluster 3; and down a piece from that are eight other gene segments that make up cluster 4. These are the players.
After the youngster grows a bit and sets his mind to getting born, one thing he wants to do is produce В cells. During the making of В cells, a funny thing happens: the DNA in the genome is rearranged, and some of it is thrown away. One segment from cluster 1 is picked out, apparently at random, and joined to one segment from cluster 2. The intervening DNA is cut out and discarded. Then a segment from cluster 3 is picked, again apparently at random, and joined to the cluster 1-2 segment. The recombining of the segments is a little bit sloppy—no what you usually expect from a cell. Because of the sloppy procedure, the coding for a few amino acids (remember, amino acids are the building blocks of proteins) can get added or lost. Once the cluster 1-2-3 segment is put together, the DNA rearrangement is over. When it's time to make an antibody, the cell makes an RNA copy of the cluster 1-2-3 combination and adds to it an RNA copy of a segment from cluster 4. Now, finally, the regions that code for contiguous protein segments are themselves in a contiguous arrangement on the RNA. How does this process explain antibody diversity? It turns out that portions of the segments from clusters 1,2, and 3 form part of the binding site—the tips of the Y. Mixing and matching different segments from the
three different clusters multiplies the number of binding sites with different shapes. For example, suppose that one segment from cluster 1 coded for a bump in the binding site, and another coded for a positive charge. And suppose that different segments from cluster 2 coded for an oily patch, a negative charge, and a deep depression, respectively. Picking one segment randomly from cluster 1 and cluster 2, you could have six possible combinations: a bump next to an oily patch, negative charge, or deep depression; or a positive charge next to an oily patch, negative charge, or deep depression. (This is essentially the same principle whereby pulling three numbers out of a hat explains the diversity of a state lottery; picking just three numbers from 0 to 9 gives a total of one thousand possible combinations.) When making an antibody heavy chain, the cell can pick one of two hundred and fifty segments from cluster 1, one of ten from cluster 2, and one of six from cluster 3. Furthermore, the sloppiness during recombination «jiggles» the segments (by crowding another amino acid into the chain, or leaving one out); this effect adds another factor of about 100 to the diversity. By mixing and matching DNA segments you get 250 × 10 × 6 × 100,which is about a million different combinations of heavy-chain sequences. Similar processes produce about ten thousand different light-chain combinations. Matching one light-chain gene to one heavy-chain gene at random in each cell gives a grand total of ten thousand times one million, or ten billion combinations! The huge number of different antibodies provides so many different binding sites that it's almost certain at least one of them will bind almost any molecule —even synthetic ones. And all of this diversity comes from a total of just about four hundred different gene segments.
The cell has other tricks to tweak upward the number of possible antibodies. One trick happens after a foreign invasion. When a cell binds to foreign material, it receives a signal to replicate; during many rounds of replication the cell «intentionally» allows a very high level of mutation in just the variable regions of the heavy- and light-chain genes. This produces variations on a winning theme. Because the parent cell coded for an antibody that already was known to bind pretty well, mutating the sequence might produce a stronger binder. In fact, studies have shown that the antibodies produced by cells late in an infection bind much more tightly to foreign molecules than antibodies produced early in an infection. This «somatic hypermutation» adds another several orders of magnitude to the diversity of possible antibodies. Remember the difference between B-cell factories and plasma factories? That oily piece of the Y that anchors the antibody in the B-cell membrane? For a plasma cell, when the RNA copy of the gene is made, the membrane segment is not copied. The segment is downstream from the rest of the gene. The DNA can be likened to a message that says «The quick brdkdjf bufjwkw nhruown fox jumps over the lapfeqmzda lfybnek sybagjufu zy dog kdjyfjdjkekiwif vmnd and eats the mnaiuw rabbit.» The final words can be left in or taken out, and the message still makes some sense.
INCH BY INCH
An antibody-diversity system requires several components to work.
The first, of course, is the genes themselves.
The second is a signal identifying the beginning and end of gene segments. In modern organisms, each segment is flanked by specific signals that tell an enzyme to come along and join the parts together. This is like a sentence that reads «The quick brcut here [fjwkw]cut hereown fox jumps over the lacut here [Ifybnek sy] cut herezy dog»—as long as the beginning and ending are present, the cell knows to keep it together.
The third component is the molecular machine that specifically recognizes the cutting signals and joins the pieces in the right order. In the absence of the machine, the parts never get cut out and joined. In the absence of the signals, it's like expecting a machine that's randomly cutting paper to make a paper doll. And, of course, in the absence of the message for the antibody itself, the other components would be pointless.
The need for minimal function reinforces the irreducible complexity of the system. Imagine you were adrift in a life raft on a stormy sea, and by chance a box floated by that contained an outboard motor. Your joy at the hope of deliverance would be short-lived if, after you affixed it to the boat, the outboard propeller turned at a rate of one revolution per day. Even if a complex system functions, the system is a failure if the level of performance is not up to snuff. The problem of the origin of antibody diversity runs headlong into the requirement for minimal function. A primitive system with only one or a few antibody molecules would be like the propeller turning at one revolution per day: not sufficient to make a difference. (More to the point, it would be as if the FBI national identification database only contained two sets of fingerprints. Out of hundreds of thousands of criminals, the FBI could only hope to catch those two.) Because the likelihood is so small for the shape of one antibody being complementary to the shape of a threatening bacterium— perhaps one in a hundred thousand or so—an animal that spent energy making five or ten antibody genes would be wasting resources that could have been invested in leaving more progeny, or building a stronger skin, or making an enzyme for excretion that would degrade RNA. To do any good, an antibody-generating system would need to generate a very large number of antibodies from the start.
THE HIT MAN
Suppose it is a thousand years ago and you live in a large compound with a group of people. Because it is near the coast, you have to worry about Viking marauders. The compound is surrounded by a strong, high wooden fence; during a raid, pots of boiling oil are poured on folks trying to climb up ladders. One strange day a traveling wizard knocks on the compound door. Opening his pack, he offers to sell you a weapon from the future. He calls it a «gun.» When the trigger is pulled, he says, the gun shoots a projectile in the direction you aim it. The gun is portable, and it could quickly be taken from one side of the compound to the other if the enemy sneakily shifted their attack. You and the other members of the compound pay the wizard two cows and four goats for the weapon. Eventually there is a raid on your compound. Boiling oil flows freely, but the raiders have a battering ram. Hearing it whack the compound gate, you stride toward the gate confidently, gun in hand. Finally the gate is smashed and the raiders pour through, screaming and waving their battle axes. You aim the gun and fire at their leader. The projectile flies through the air and sticks to the Viking chieftains nose. On the barrel of the gun, in letters you cannot read, is the inscription «Acme Toy Dart Gun.» The chieftain stops, stares at you, and begins to grin as your smile dissolves. He and his friends rush at you; fortunately, you are reincarnated as a biochemist in the twentieth century. Antibodies are like toy darts: they harm no one. Like a «Condemned» sign posted on an old house or an orange «X» painted on a tree to be removed, antibodies are only signals to other systems to destroy the marked object. It is surprising to think that after the body has gone to all the trouble to develop a complex system to generate antibody diversity, and after it has laboriously picked a few cells by the roundabout process of clonal selection, it is still virtually helpless against the onslaught of invaders.
Much of the actual killing of foreign cells that are marked by antibodies is done by the «complement» system, which is called this because it complements the action of antibodies in getting rid of invaders.
The pathway is remarkably complex (Figure 6-3);
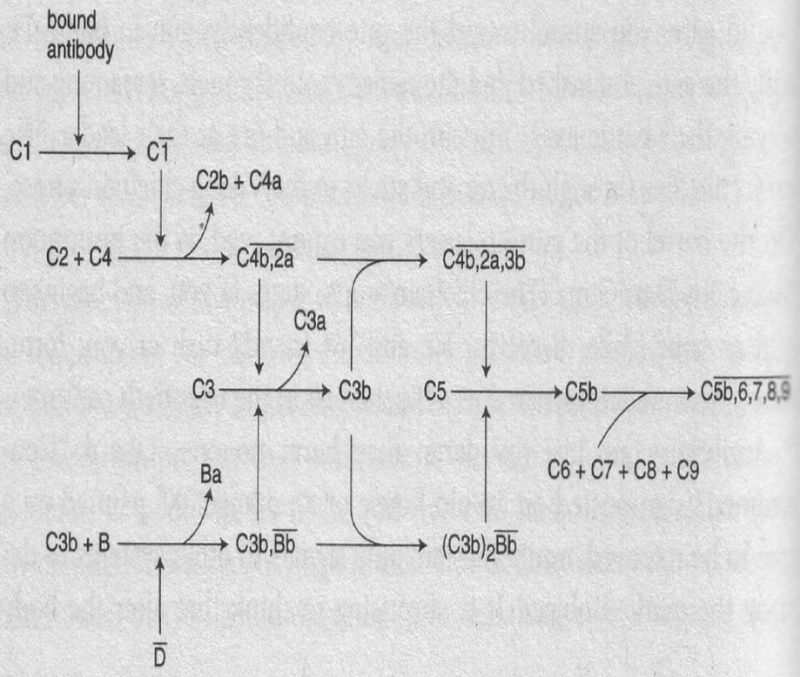
FIGURE 6-3
in many ways, it is similar to the blood-clotting cascade. It consists of about 20 kinds of proteins that form two related pathways, called the classical pathway and the alternative pathway. The classical pathway starts when a large aggregate of proteins, called C1, binds to an antibody that is itself bound to the surface of a foreign cell. It is crucial that the C1 complex recognize only bound antibody; if C1 attached itself to antibody that was floating around in the bloodstream, then all of the C1 would be sopped up and unavailable for action against enemies. Or, if C1 bound to the membrane-attached antibodies of В cells, it would initiate reactions that ultimately would end up killing good cells. C1 is made up of 22 protein chains. These can be divided into three groups. The first is called C1q. It contains six copies of three different types of proteins, for a total of 18. The other two groups are called C1r and С1s. They both have two copies each of different proteins. The three different types of proteins in C1q all begin with a special amino-acid sequence that resembles the sequence of the skin protein collagen. The sequence allows the tails of the three types of C1q proteins to wrap around each other like braids. This arrangement holds one of each type of protein in a minicomplex. The remainder of the protein chains then fold up into complex, globular shapes at the top of the braid. Six of the minicomplexes then come together. The six braids stick to each other lengthwise to create a central stalk, out of which protrude six heads. Pictures of C1q taken with an electron microscope show something resembling a hydra-headed monster. (Other people have likened it to a bouquet of tulips, but I like more dramatic images.) The C1q heads attach to the antibody-foreign cell complex. At least two of the heads have to be attached before the pathway is initiated. Once they stick, something in C1q changes, and the change in C1q causes C1r and С1s to bind more tightly to C1q. When this happens C1r cuts itself (headline: Dog bites dog!) to give C1r. («Activated» proteins are designated by an upper bar over the number and lower case letter.) C1r then is able to cut С1s to yield С1s. After С1s is cleaved, we still have a long way to go before the work of destroying the invading cell is finished. The proteins of C1 are collectively called the «recognition unit.» The next group of proteins (named C2, C3, and C4) is called the «activation unit.» Unlike the recognition unit, the activation unit is not already together in one piece; it has to be assembled. The first step in forming the activation unit is the cleavage of C4 by C1s. When C4 is cut by C1s, a very reactive group that was inside one piece (C4b) is exposed to the surroundings. If the group is close to a membrane, it can chemically react with it. The attachment of C4b is necessary so the rest of the proteins in the activation unit can have an anchor to hold them close to the invader. In contrast, if C4b is pointed in the wrong direction or is floating around in solution, then the reactive group quickly decays without attaching to the correct membrane. After C4b has attached itself to the target membrane, in association with C1s it cleaves C2 into two pieces. The larger piece, C2a, remains stuck to C4b to yield C4b,2a, also known as «C3 convertase.»

C3 convertase has to act quickly, or it falls apart and C2a floats away. If a molecule of C3 is in the vicinity, C3 convertase cleaves it into two pieces. C3b sticks to C3 convertase to form C4b,2a,3b, which is also called «C5 convertase.» The final reaction of the activation unit is the cleavage of C5 into two fragments. At this point the system is finally ready to stick a knife in the invader. One of the pieces of C5 sticks to C6 and C7. This structure has the remarkable property of being able to insert itself into a cell membrane. C5b,6,7 then binds to a molecule of C8 and a variable number (from one to eighteen) of molecules of C9
adds to it. The proteins, however, do not form an undifferentiated glob. Rather, they organize themselves into a tubular form that punches a hole in the membrane of the invading bacterial cell. Because the insides of cells are very concentrated solutions, osmotic pressure causes water to rush in. The in-rushing water swells the bacterial cell till it bursts. There is an alternative pathway for the activation of the membrane-attack complex that can act quickly after infection, not needing to wait for the production of specific antibodies. In the alternative pathway a small amount of C3b, which apparently is produced continuously in low amounts, binds with a protein called factor B. C3b,B can then be cut by another protein, factor D, to give C3b,Bb. This can now act as a C3 convertase. When more C3b is made, a second molecule of C3b can attach to yield (C3b) Bb. Remarkably, this is now a C5 convertase, which produces 2 C5b, which then goes on to start the formation of the membrane-attack complex in the way described above for the first pathway.
C3b is a dangerous protein to have floating around, since it can activate the destructive end of the complement pathway. In order to minimize random damage, two proteins (factors I and H), search out, stick to, and destroy C3b in solution. But if C3b is on the surface of a cell, then another protein (properdin), binds to and protects C3b from degradation so that it can do its job. How does C3b target foreign cells in the absence of antibodies? C3b is effective only if it sticks to the surface of a cell. The chemical reaction by which it does so goes faster in the presence of the molecules typically found on the surface of many bacteria and viruses.
Like the blood-clotting pathway, the complement pathway is a cascade. Inevitably, in both cases one encounters the same problems trying to imagine their gradual production. It is not the final activity of a cascade that is the problem. The formation of a hole in a membrane does not necessarily require several different components; one killer protein could conceivably do the job. Nor does the formation of a protein aggregate, such as in blood clotting, necessarily require multiple components; under the right conditions, any protein will aggregate. (The particular shapes of the complement hole-complex and fibrin aggregate, however, are particularly suited to the jobs they do and need to be explained.) And as we saw in Chapter 4, a telephone pole by itself could bop Foghorn Leghorn. It is the control systems that are the problem. At each control point both the regulatory protein and the masked protein that it activates have to be present from the beginning. If C5b were present, the rest of the cascade would immediately be touched off; but if C5 were present with nothing to activate it, then the whole pathway would always be shut off. If C3b were present, the rest of the cascade would immediately be touched off; but if C3 were present with nothing to activate it, then the whole pathway would always be shut off. Even if one imagines a much shortened pathway (where, say, Cls directly cuts C5), insertion of additional control points into the middle of the cascade runs into the same problem: the irreducible complexity of the switches.
In addition to the generic problems of setting up a cascade, the complement pathway shares another problem with the blood-clotting cascade: attachment of proteins to membranes is crucial. Several clotting factors must first be modified to synthesize Gla residues so that they could stick to a membrane. In the complement pathway, both C3 and C4 have unusual, highly reactive internal groups that chemically attach to the membrane after the proteins are cleaved by other factors. These special features have to be available before the pathway is functional, adding a further severe barrier to their gradual development. Numerous little features of the complement system are stumbling blocks to gradual development. Let's consider some subtle characteristics of just the C1 system. The three types of proteins in C1q braid around each other, but do not braid with themselves. If they did, then the ratio of different types of chains in the complex would be changed, and there would be a much smaller chance of getting the real C1q complex with six copies of three different chains. If the binding of C1q to the antibody-foreign cell did not trigger C1r's self-scission, then the cascade would be stopped in its tracks. Conversely, if C1r cut itself before C1q bound to the antibody complex, then the cascade would be prematurely triggered. And so on.
The proper functioning of the immune system is a prerequisite for health. Major illnesses such as cancer and AIDS have either their cause or their cure, or both, in the vagaries of the system. Because of its impact on public health, the immune system is a subject of intense interest. Thousands of research laboratories around the world work on various aspects of the immune system. Their efforts have already saved many lives and promise to save many more in the future.
Although great strides have been made in understanding how the immune system works, we remain ignorant of how it came to be. None of the questions raised has been answered by any of the thousands of scientists in the field; few have even asked the questions. A search of the immunological literature shows ongoing work in comparative immunology (the study of immune systems from various species). But that work, valuable though it is, does not address in molecular detail the question of how immune systems originated. Perhaps the best efforts at doing that so far have been in two short papers. The first, by Nobel laureate David Baltimore and two other prominent scientists, is tantalizingly entitled «Molecular Evolution of the Vertebrate Immune System.» But it's hard to live up to such a title in just two pages. The authors point out that for any organism to have an immune system akin to that seen in mammals, the minimally required molecules are the antigen receptors (immunoglobulin and TCR), the antigen presentation molecules (MHC), and the gene rearranging proteins. (Immunoglobulins are antibodies. TCR molecules are akin to antibodies.) The authors then argue that sharks, which are very distantly related to mammals, appear to have all three components. But it's one thing to say an organism has a completed, functioning system, and another to say how the system developed. The authors certainly realize this. They note that immunoglobulin and TCR genes both require RAG proteins for rearrangement. On the other hand, RAG proteins require specific recombination signals to rearrange immunoglobulin and TCR genes. (RAG is the component that rearranges the genes.) They make a valiant stab at accounting for the components, but in the end, it is a hop in the box with Calvin and Hobbes. The authors speculate that a gene from a bacterium might have luckily been transferred to an animal. Luckily, the protein coded by the gene could itself rearrange genes; and luckily, in the animal's DNA there were signals that were near antibody genes; and so on. In the final analysis the authors identify key problems with gradualistic evolution of the immune system, but their proffered solutions are really just a disguised shrug of their shoulders.
Another paper that gamely tries to account for a piece of the immune system is entitled «Evolution of the Complement System.»6 Like the paper discussed above, it is very short and is a commentary article—in other words, not a research article. The authors make some imaginative guesses about what might come first and second, but inevitably they join Russell Doolittle in proposing unexplained proteins that are «unleashed» and «spring forth» («At some point a critical gene fusion created a protease with a binding site for the primitive C3b»; «Evolution of the other alternative pathway components further improved the amplification and specificity»; and «C2, created by the duplication of the factor В gene, would then have allowed further divergence and specialization of the two pathways»). No quantitative calculations appear in the paper. Nor does an acknowledgment that gene duplications would not immediately make a new protein. Nor does any worry about a lack of controls to regulate the pathway. But then, it would be hard to fit those concerns in the four paragraphs of the paper that deal with molecular mechanisms.
There are other papers and books that discuss the evolution of the immune system.7 Most of them, however, are at the level of cell biology and thus unconcerned with detailed molecular mechanisms, or else they are concerned simply with comparison of DNA or protein sequences. Comparing sequences might be a good way to study relatedness, but the results can't tell us anything about the mechanism that first produced the systems.
We can look high or we can look low, in books or in journals, but the result is the same. The scientific literature has no answers to the question of the origin of the immune system.
I have looked at three features of the immune system—clonal selection, antibody diversity, and the complement system—and demonstrated that each individually poses massive challenges to a putative step-by-step evolution. But showing that the parts can't be built step by step only tells part of the story, because the parts interact with each other. Just as a car without steering, or a battery, or a carburetor isn't going to do you much good, an animal that has a clonal selection system won't get much benefit out of it if there is no way to generate antibody diversity. A large repertoire of antibodies won't do much good if there is no system to kill invaders. A system to kill invaders won't do much good if there's no way to identify them. At each step we are stopped not only by local system problems, but also by requirements of the integrated system.
clonal selection
antibody diversity
system to kill invaders
a way to identify them
https://reasonandscience.catsboard.com/t2322-the-immune-system-and-irreducible-complexity
Darwins black box, page 97
Although great strides have been made in understanding how the immune system works, we remain ignorant of how it came to be. None of the questions raised has been answered by any of the thousands of scientists in the field; few have even asked the questions. A search of the immunological literature shows ongoing work in comparative immunology (the study of immune systems from various species). But that work, valuable though it is, does not address in molecular detail the question of how immune systems originated.
Perhaps the best efforts at doing that so far have been in two short papers. The first, by Nobel laureate David Baltimore and two other prominent scientists, is tantalizingly entitled «Molecular Evolution of the Vertebrate Immune System.» But it's hard to live up to such a title in just two pages. The authors point out that for any organism to have an immune system akin to that seen in mammals, the minimally required molecules are the antigen receptors (immunoglobulin and TCR), the antigen presentation molecules (MHC), and the gene rearranging proteins. (Immunoglobulins are antibodies. TCR molecules are akin to antibodies.) The authors then argue that sharks, which are very distantly related to mammals, appear to have all three components.
But it's one thing to say an organism has a completed, functioning system, and another to say how the system developed. The authors certainly realize this. They note that immunoglobulin and TCR genes both require RAG proteins for rearrangement. On the other hand, RAG proteins require specific recombination signals to rearrange immunoglobulin and TCR genes. (RAG is the component that rearranges the genes.) They make a valiant stab at accounting for the components, but in the end, it is a hop in the box with Calvin and Hobbes. The authors speculate that a gene from a bacterium might have luckily been transferred to an animal. Luckily, the protein coded by the gene could itself rearrange genes; and luckily, in the animal's DNA there were signals that were near antibody genes; and so on. In the final analysis the authors identify key problems with gradualistic evolution of the immune system, but their proffered solutions are really just a disguised shrug of their shoulders.
I have looked at three features of the immune system—clonal selection, antibody diversity, and the complement system—and demonstrated that each individually poses massive challenges to a putative step-by-step evolution. But showing that the parts can't be built step by step only tells part of the story, because the parts interact with each other. Just as a car without steering, or a battery, or a carburetor isn't going to do you much good, an animal that has a clonal selection system won't get much benefit out of it if there is no way to generate antibody diversity. A large repertoire of antibodies won't do much good if there is no system to kill invaders. A system to kill invaders won't do much good if there's no way to identify them. At each step we are stopped not only by local system problems, but also by requirements of the integrated system.
THE RIGHT STUFF
When a microscopic invader breaches the outer defenses of the body the immune system swings into action. This happens automatically. The molecular systems of the body, like the Star Wars anti-missile system that the military once planned, are robots designed to run on autopilot. Since the defense is automated, every step has to be accounted for by some mechanism. The first problem that the automated defense system has is how to recognize an invader. Bacterial cells have to be distinguished from blood cells; viruses have to be distinguished from connective tissue. Unlike us, the immune system can't see, so it has to rely initially on something akin to a sense of touch. Antibodies are the «fingers» of the blind immune system—they allow it to distinguish a foreign invader from the body itself. Antibodies are formed by an aggregation of four chains of amino acids (Figure 6-1):

two identical light chains, and two identical heavy chains. The heavy chains are about twice as big as the light chains. In the cell, the four chains make a complex that resembles the letter Y. Because the two heavy chains are the same and the two light chains are the same, the Y is symmetrical: if you took a knife and cut it down the middle you'd get identical halves, with one heavy and one light chain in each half. At the end of each pronged tip of the Y there is a depression (called a binding site). Lining the binding site are portions of both the light chain and the heavy chain. Binding sites come in a large variety of shapes. One antibody might have a binding site with a piece jutting up here, a hole over there, and an oily patch on the edge. A second antibody might have a positive charge on the left, a crevice in the middle, and a bump on the right.
If the shape of a binding site just happens to be exactly complementary to the shape of a molecule on the surface of an invading virus or bacterium, then the antibody will bind to that molecule. To get a feel for it, imagine a household object with a depression in it and a few knobs poking up out of the depression. My youngest daughter has a doll wagon with front and back seats—something like that will do nicely. Now take the wagon/object, go around the house, and see how many other articles will fit snugly into the depression, filling both the front seat and the back seat without leaving any spaces. If you find even one, you're luckier than I am. Nothing in my house fit snugly in the wagon, and neither did anything in my office or laboratory. I imagine there's some object out in the world with a shape complementary to the wagon's, but I haven't found it yet. The body has a similar problem: the odds of any given antibody binding to any given invader are pretty slim. To make sure that at least one kind of antibody is available for each attacker, we make billions to trillions of them. Usually, for any particular invader; it takes 100,000 to find one antibody that works. When bacteria invade the body, they multiply.
By the time an antibody binds to a bacterium there may be many, many copies of the bug floating around. Against this Trojan horse that breeds, the body has 100,000 guns, but only one works. One handgun isn't going to do much good against a horde; somehow reinforcements have to be brought in. There's a way to do this, but first I have to back up and explain a bit more about where antibodies come from. There are billions of different kinds of antibodies. Each kind of antibody is made in a separate cell. The cells that make antibodies are called В cells, which is easy to remember because they are produced in the bone marrow. When а В cell is first born, mechanisms inside of it randomly choose one of the many antibody genes that are encoded in its DNA. That gene is said to be «turned on»; all other antibody genes are «turned off.» So the cell produces only one kind of antibody, with one kind of binding site. The next cell that's made will in all likelihood have a different antibody gene turned on, so it will make a different protein with a different binding site.
The principle, then, is one cell, one type of antibody. Once a cell commits to making its antibody, you might think that the antibody would leave the cell so it could patrol the body. But if the contents of all В cells were dumped out into the body, there would be no way to tell which cell the antibody came from. The cell is the factory that makes the particular type of antibody; if the antibody finds a bacterium, we need to tell the cell to send us reinforcements. But with this hypothetical setup, we can't get a message back. Fortunately, the body is smarter than that. When а В cell first makes its antibody, the antibody anchors in the cell membrane with the prongs of the Y sticking out (Figure 6-2).

The cell does this trick by using the gene for the normal antibody, and also using a little piece of a gene that codes for an oily tail on the protein. Since the membrane is oily, too, the piece sticks in the membrane. This step is critical, because now the binding site of the antibody is attached to its factory. The entire В cell factory patrols the body; when a foreign invader enters, the antibody-with-attached-cell binds. Now we have the factory close at hand to the invaders. If the cell could be signaled to make more of the antibody, then the fight would be helped by reinforcements. Fortunately, there is a way to send a signal; unfortunately, it's pretty convoluted. When an antibody on а В cell binds to a foreign molecule it triggers a complex mechanism to swallow the invader: in effect, the munitions factory takes a hostage. The antibody then breaks off a piece of membrane to make a little vesicle—a self-made taxicab. In this taxi, the hostage is brought into the B-cell factory. Inside the cell (still in the cab) the foreign protein is chopped up, and a piece of the foreign protein sticks to another
protein (called an MHC protein). The cab then returns to the membrane of the cell. Outside the factory, along comes another cell (called a helper T cell). The helper T cell binds to the В cell, which is «presenting» the chopped-up piece of invader (the foreign fragment in the MHC protein) for the T cell's consideration. If the fit is just right, it causes the helper T cell to secrete a substance called interleukin. Interleukin is like a message from the Department of Defense to the munitions factory. By binding to another protein on the surface of the В cell, the interleukin sets off a chain of events that sends a message to the nucleus of the В cell. The message is: grow!
The В cell begins to reproduce at a rapid rate. T cells continue to secrete interleukin if they are bound to а В cell. Eventually the growing B-cell factory produces a series of spinoff factories in the form of specialized cells called 'plasma cells.' Instead of producing a form of the antibody that sticks in the membrane, plasma cells leave off the last oily piece of the protein. Now free antibody is extruded in large amounts into the extracellular fluid. The switch is critical. If the new plasma-cell factories were like the old B-cell factory, the antibodies would all be confined to quarters and would be much less effective at inhibiting the invaders.
STEP BY STEP
Could this system have evolved step-by-step? Consider the vast pool billions to trillions of factory В cells. The process of picking the right cell out of a mixture of antibody-producing cells is called clonal selection. Clonal selection is an elegant way to mount a specific response in great numbers to a wide variety of possible foreign invaders. The process depends on a large number of steps, some of which I have not discussed yet. Leaving those aside for now, let's ask what the minimum requirements are for a clonal selection system, and if those minimum requirements could be produced step-by-step. The key to the system is the physical connection of the binding ability of the protein with the genetic information for the protein. Theoretically this could be accomplished by making an antibody where the tail of the Y bound to the DNA that coded for the protein. In real life, however, such a setup wouldn't work. The protein might be connected to its genetic information, but because the cell is surrounded by a membrane, the antibody would never come in contact with the foreign material, which is floating around outside the cell. A system where both the antibody and its attached gene were exported from the cell would overcome that problem, only to run into a different one: outside the cell there would be no cellular machinery to translate the DNA message into more protein. Anchoring the antibody in the membrane is a good solution to the problem; now the antibody can mix it up with a foreign cell and still be near its DNA. But although the antibody can bind the foreign material without floating away from the cell, it does not have direct physical contact with the DNA. Since the protein and DNA are blind, there must be a way to get a message from one to the other. Just for now, for the sake of argument, let's forget about the tortuous way that the message of binding actually gets to the B-cell nucleus (requiring the taxicab, ingestion, MHC, helper T cells, interleukin, and so on).
Instead let's imagine a simpler system where there's only one other protein. Let's say that when the antibody binds to a foreign molecule, something happens that attracts some other protein—a messenger to take word of a hostage to the factory nucleus. Maybe when the hostage is first found, the shape of the antibody changes, perhaps pulling up a little on the antibody's tail. Perhaps part of the antibody's tail sticks into the inside of the cell, which is what triggers the messenger protein. The change in the tail could cause the messenger protein to scuttle into the nucleus and bind to the DNA at a particular point. Binding to the right place on the DNA is what causes the cell to start growing and to start producing antibody without the oily tail—antibody that gets sent out of the cell to fight the invasion. Even in such a simplified scheme, we are left with three critical ingredients:
(1) the membrane-bound form of the antibody;
(2) the messenger; and
(3) the exported form of the antibody.
If any of these components is missing, the system fails to function. If there is no antibody in the membrane, then there's no way to connect a successful antibody that binds a foreign invader to the cell containing the genetic information. If there is no exported form of the antibody, then when the signal is received there is nothing to send out into the world to fight. If there is no messenger protein, then there is no connection between binding the membrane antibody and turning on the right gene (making the system about as useful as a doorbell whose wires had been cut). A cell hopefully trying to evolve such a system in gradual Darwinian steps would be in a quandary. What should it do first? Secreting a little bit of antibody into the great outdoors is a waste of resources if there's no way to tell if it's doing any good. Ditto for making a membrane-bound antibody. And why make a messenger protein first if there is nobody to give it a message, and nobody to receive the message if it did get one? We are led inexorably to the conclusion that even this greatly simplified clonal selection could not have come about in gradual steps.
Even at this simplified level, then, all three ingredients had to evolve simultaneously. Each of these three items—the fixed antibody, the messenger protein, and the loose antibodies—had to be produced by a separate historical event, perhaps by a coordinated series of mutations changing preexisting proteins that were doing other chores into the components of the antibody system. Darwin's small steps have become a series of wildly unlikely leaps.
Yet our analysis overlooked many complexities: How does the cell switch from putting the extra oily piece on the membrane to not putting it on? The message system then is fantastically more complicated than our simplified version. Ingestion of the protein, chopping it up, presenting it to the outside on an MHC protein, specific recognition of the МНС/fragment by a helper T cell, secretion of interleukin, binding of interleukin to the В cell, sending the signal that interleukin has bound into the nucleus— the prospect of devising a step-by-step pathway for the origin of the system is enough to make strong men blanch.Factories float around in huge numbers, poised to deliver antibodies that can stick to an invader with virtually any shape. But how does the body make all those billions of differently shaped antibodies? It turns out that there is an elegant trick for making very many different antibodies without requiring enormous quantities of genetic material to code for the proteins.
Again, don't be concerned if the details quickly slip your mind; my purpose here is just to help you appreciate the complexity of the immune system. It took a fascinating discovery to lead scientists to puzzle out the full complexity of the immune system. The discovery started with a potentially cruel, but necessary, experiment. Just to see what would happen, chemists made some small molecules that do not occur in nature and then attached them to a protein. When the protein carrying the synthetic molecules was injected into a rabbit, the scientists were astonished to find that, yes, the rabbit made antibodies that bound tightly to the synthetic molecule. How could this be? Neither the rabbit nor its ancestors ever met the synthetic molecule, so how did it know how to make antibodies against it? Why should it recognize a molecule it had never seen before?
The puzzle of «antibody diversity» intrigued scientists studying immunology. Several ideas were floated as possible explanations. Proteins were known to be flexible molecules, and antibodies are proteins. So maybe when a new molecule is injected into the body an antibody wraps around it, molds itself to that shape, and then somehow freezes in that configuration. Or maybe, because defense is so vitally important, the DNA of organisms contains a vast number of genes for antibodies with many different shapes—enough to allow them to recognize things they hadn't seen yet. But such a huge number of antibodies would take up more than the available coding space in the DNA. So maybe there were only a few antibodies, and when the cell divided, maybe there was some way to make a lot of mutations in just the areas coding for the binding sites of the antibodies. That way each new В cell in the body could carry different mutations, coding for an antibody different from all other В cells. Or maybe the answer was a combination of these, or maybe it involved something completely new.
The answer to the problem of antibody diversity had to await an astonishing discovery: a gene coding for a protein didn't always have to be a continuous segment of DNA—it could be interrupted. If we compare a gene to a sentence, it was as if a protein's code, «The quick brown fox jumps over the lazy dog» could be altered (without destroying the protein) to read «The quick brdkdjf bufjwkw nhruown fox jumps over the lapfeqmzda lfybnek sybagjufu zy dog.» The sensible DNA message was broken up by tracts of nonsense letters that somehow were not included in the protein. Further work showed that for most genes, corrections would be made— splicing out the nonsense—after an RNA copy is made of a DNA gene.
Even with «interrupted» DNA, an edited and corrected message in RNA could be used by the cell's machinery to make the correct protein. Even more surprisingly, for antibody genes the DNA itself can also be spliced. In other words, DNA that is inherited can be altered. Amazing! Splicing and rearrangement of DNA play a large role in explaining the great number of antibodies that the body can produce.
The following is a brief description of work that has taken many investigators many years to accomplish; because of their efforts, the riddle of antibody diversity is solved.
At conception there are a number of gene pieces in the fertilized cell that contribute to making antibodies. The genes are arranged into clusters that I will simply call cluster 1, cluster 2, and so forth. In humans there are approximately 250 gene segments in cluster 1; a ways down the DNA from cluster 1 are ten gene segments that form cluster 2; further on down the DNA road are a group of six segments that comprise cluster 3; and down a piece from that are eight other gene segments that make up cluster 4. These are the players.
After the youngster grows a bit and sets his mind to getting born, one thing he wants to do is produce В cells. During the making of В cells, a funny thing happens: the DNA in the genome is rearranged, and some of it is thrown away. One segment from cluster 1 is picked out, apparently at random, and joined to one segment from cluster 2. The intervening DNA is cut out and discarded. Then a segment from cluster 3 is picked, again apparently at random, and joined to the cluster 1-2 segment. The recombining of the segments is a little bit sloppy—no what you usually expect from a cell. Because of the sloppy procedure, the coding for a few amino acids (remember, amino acids are the building blocks of proteins) can get added or lost. Once the cluster 1-2-3 segment is put together, the DNA rearrangement is over. When it's time to make an antibody, the cell makes an RNA copy of the cluster 1-2-3 combination and adds to it an RNA copy of a segment from cluster 4. Now, finally, the regions that code for contiguous protein segments are themselves in a contiguous arrangement on the RNA. How does this process explain antibody diversity? It turns out that portions of the segments from clusters 1,2, and 3 form part of the binding site—the tips of the Y. Mixing and matching different segments from the
three different clusters multiplies the number of binding sites with different shapes. For example, suppose that one segment from cluster 1 coded for a bump in the binding site, and another coded for a positive charge. And suppose that different segments from cluster 2 coded for an oily patch, a negative charge, and a deep depression, respectively. Picking one segment randomly from cluster 1 and cluster 2, you could have six possible combinations: a bump next to an oily patch, negative charge, or deep depression; or a positive charge next to an oily patch, negative charge, or deep depression. (This is essentially the same principle whereby pulling three numbers out of a hat explains the diversity of a state lottery; picking just three numbers from 0 to 9 gives a total of one thousand possible combinations.) When making an antibody heavy chain, the cell can pick one of two hundred and fifty segments from cluster 1, one of ten from cluster 2, and one of six from cluster 3. Furthermore, the sloppiness during recombination «jiggles» the segments (by crowding another amino acid into the chain, or leaving one out); this effect adds another factor of about 100 to the diversity. By mixing and matching DNA segments you get 250 × 10 × 6 × 100,which is about a million different combinations of heavy-chain sequences. Similar processes produce about ten thousand different light-chain combinations. Matching one light-chain gene to one heavy-chain gene at random in each cell gives a grand total of ten thousand times one million, or ten billion combinations! The huge number of different antibodies provides so many different binding sites that it's almost certain at least one of them will bind almost any molecule —even synthetic ones. And all of this diversity comes from a total of just about four hundred different gene segments.
The cell has other tricks to tweak upward the number of possible antibodies. One trick happens after a foreign invasion. When a cell binds to foreign material, it receives a signal to replicate; during many rounds of replication the cell «intentionally» allows a very high level of mutation in just the variable regions of the heavy- and light-chain genes. This produces variations on a winning theme. Because the parent cell coded for an antibody that already was known to bind pretty well, mutating the sequence might produce a stronger binder. In fact, studies have shown that the antibodies produced by cells late in an infection bind much more tightly to foreign molecules than antibodies produced early in an infection. This «somatic hypermutation» adds another several orders of magnitude to the diversity of possible antibodies. Remember the difference between B-cell factories and plasma factories? That oily piece of the Y that anchors the antibody in the B-cell membrane? For a plasma cell, when the RNA copy of the gene is made, the membrane segment is not copied. The segment is downstream from the rest of the gene. The DNA can be likened to a message that says «The quick brdkdjf bufjwkw nhruown fox jumps over the lapfeqmzda lfybnek sybagjufu zy dog kdjyfjdjkekiwif vmnd and eats the mnaiuw rabbit.» The final words can be left in or taken out, and the message still makes some sense.
INCH BY INCH
An antibody-diversity system requires several components to work.
The first, of course, is the genes themselves.
The second is a signal identifying the beginning and end of gene segments. In modern organisms, each segment is flanked by specific signals that tell an enzyme to come along and join the parts together. This is like a sentence that reads «The quick brcut here [fjwkw]cut hereown fox jumps over the lacut here [Ifybnek sy] cut herezy dog»—as long as the beginning and ending are present, the cell knows to keep it together.
The third component is the molecular machine that specifically recognizes the cutting signals and joins the pieces in the right order. In the absence of the machine, the parts never get cut out and joined. In the absence of the signals, it's like expecting a machine that's randomly cutting paper to make a paper doll. And, of course, in the absence of the message for the antibody itself, the other components would be pointless.
The need for minimal function reinforces the irreducible complexity of the system. Imagine you were adrift in a life raft on a stormy sea, and by chance a box floated by that contained an outboard motor. Your joy at the hope of deliverance would be short-lived if, after you affixed it to the boat, the outboard propeller turned at a rate of one revolution per day. Even if a complex system functions, the system is a failure if the level of performance is not up to snuff. The problem of the origin of antibody diversity runs headlong into the requirement for minimal function. A primitive system with only one or a few antibody molecules would be like the propeller turning at one revolution per day: not sufficient to make a difference. (More to the point, it would be as if the FBI national identification database only contained two sets of fingerprints. Out of hundreds of thousands of criminals, the FBI could only hope to catch those two.) Because the likelihood is so small for the shape of one antibody being complementary to the shape of a threatening bacterium— perhaps one in a hundred thousand or so—an animal that spent energy making five or ten antibody genes would be wasting resources that could have been invested in leaving more progeny, or building a stronger skin, or making an enzyme for excretion that would degrade RNA. To do any good, an antibody-generating system would need to generate a very large number of antibodies from the start.
THE HIT MAN
Suppose it is a thousand years ago and you live in a large compound with a group of people. Because it is near the coast, you have to worry about Viking marauders. The compound is surrounded by a strong, high wooden fence; during a raid, pots of boiling oil are poured on folks trying to climb up ladders. One strange day a traveling wizard knocks on the compound door. Opening his pack, he offers to sell you a weapon from the future. He calls it a «gun.» When the trigger is pulled, he says, the gun shoots a projectile in the direction you aim it. The gun is portable, and it could quickly be taken from one side of the compound to the other if the enemy sneakily shifted their attack. You and the other members of the compound pay the wizard two cows and four goats for the weapon. Eventually there is a raid on your compound. Boiling oil flows freely, but the raiders have a battering ram. Hearing it whack the compound gate, you stride toward the gate confidently, gun in hand. Finally the gate is smashed and the raiders pour through, screaming and waving their battle axes. You aim the gun and fire at their leader. The projectile flies through the air and sticks to the Viking chieftains nose. On the barrel of the gun, in letters you cannot read, is the inscription «Acme Toy Dart Gun.» The chieftain stops, stares at you, and begins to grin as your smile dissolves. He and his friends rush at you; fortunately, you are reincarnated as a biochemist in the twentieth century. Antibodies are like toy darts: they harm no one. Like a «Condemned» sign posted on an old house or an orange «X» painted on a tree to be removed, antibodies are only signals to other systems to destroy the marked object. It is surprising to think that after the body has gone to all the trouble to develop a complex system to generate antibody diversity, and after it has laboriously picked a few cells by the roundabout process of clonal selection, it is still virtually helpless against the onslaught of invaders.
Much of the actual killing of foreign cells that are marked by antibodies is done by the «complement» system, which is called this because it complements the action of antibodies in getting rid of invaders.
The pathway is remarkably complex (Figure 6-3);

FIGURE 6-3
in many ways, it is similar to the blood-clotting cascade. It consists of about 20 kinds of proteins that form two related pathways, called the classical pathway and the alternative pathway. The classical pathway starts when a large aggregate of proteins, called C1, binds to an antibody that is itself bound to the surface of a foreign cell. It is crucial that the C1 complex recognize only bound antibody; if C1 attached itself to antibody that was floating around in the bloodstream, then all of the C1 would be sopped up and unavailable for action against enemies. Or, if C1 bound to the membrane-attached antibodies of В cells, it would initiate reactions that ultimately would end up killing good cells. C1 is made up of 22 protein chains. These can be divided into three groups. The first is called C1q. It contains six copies of three different types of proteins, for a total of 18. The other two groups are called C1r and С1s. They both have two copies each of different proteins. The three different types of proteins in C1q all begin with a special amino-acid sequence that resembles the sequence of the skin protein collagen. The sequence allows the tails of the three types of C1q proteins to wrap around each other like braids. This arrangement holds one of each type of protein in a minicomplex. The remainder of the protein chains then fold up into complex, globular shapes at the top of the braid. Six of the minicomplexes then come together. The six braids stick to each other lengthwise to create a central stalk, out of which protrude six heads. Pictures of C1q taken with an electron microscope show something resembling a hydra-headed monster. (Other people have likened it to a bouquet of tulips, but I like more dramatic images.) The C1q heads attach to the antibody-foreign cell complex. At least two of the heads have to be attached before the pathway is initiated. Once they stick, something in C1q changes, and the change in C1q causes C1r and С1s to bind more tightly to C1q. When this happens C1r cuts itself (headline: Dog bites dog!) to give C1r. («Activated» proteins are designated by an upper bar over the number and lower case letter.) C1r then is able to cut С1s to yield С1s. After С1s is cleaved, we still have a long way to go before the work of destroying the invading cell is finished. The proteins of C1 are collectively called the «recognition unit.» The next group of proteins (named C2, C3, and C4) is called the «activation unit.» Unlike the recognition unit, the activation unit is not already together in one piece; it has to be assembled. The first step in forming the activation unit is the cleavage of C4 by C1s. When C4 is cut by C1s, a very reactive group that was inside one piece (C4b) is exposed to the surroundings. If the group is close to a membrane, it can chemically react with it. The attachment of C4b is necessary so the rest of the proteins in the activation unit can have an anchor to hold them close to the invader. In contrast, if C4b is pointed in the wrong direction or is floating around in solution, then the reactive group quickly decays without attaching to the correct membrane. After C4b has attached itself to the target membrane, in association with C1s it cleaves C2 into two pieces. The larger piece, C2a, remains stuck to C4b to yield C4b,2a, also known as «C3 convertase.»
C3 convertase has to act quickly, or it falls apart and C2a floats away. If a molecule of C3 is in the vicinity, C3 convertase cleaves it into two pieces. C3b sticks to C3 convertase to form C4b,2a,3b, which is also called «C5 convertase.» The final reaction of the activation unit is the cleavage of C5 into two fragments. At this point the system is finally ready to stick a knife in the invader. One of the pieces of C5 sticks to C6 and C7. This structure has the remarkable property of being able to insert itself into a cell membrane. C5b,6,7 then binds to a molecule of C8 and a variable number (from one to eighteen) of molecules of C9
adds to it. The proteins, however, do not form an undifferentiated glob. Rather, they organize themselves into a tubular form that punches a hole in the membrane of the invading bacterial cell. Because the insides of cells are very concentrated solutions, osmotic pressure causes water to rush in. The in-rushing water swells the bacterial cell till it bursts. There is an alternative pathway for the activation of the membrane-attack complex that can act quickly after infection, not needing to wait for the production of specific antibodies. In the alternative pathway a small amount of C3b, which apparently is produced continuously in low amounts, binds with a protein called factor B. C3b,B can then be cut by another protein, factor D, to give C3b,Bb. This can now act as a C3 convertase. When more C3b is made, a second molecule of C3b can attach to yield (C3b) Bb. Remarkably, this is now a C5 convertase, which produces 2 C5b, which then goes on to start the formation of the membrane-attack complex in the way described above for the first pathway.
C3b is a dangerous protein to have floating around, since it can activate the destructive end of the complement pathway. In order to minimize random damage, two proteins (factors I and H), search out, stick to, and destroy C3b in solution. But if C3b is on the surface of a cell, then another protein (properdin), binds to and protects C3b from degradation so that it can do its job. How does C3b target foreign cells in the absence of antibodies? C3b is effective only if it sticks to the surface of a cell. The chemical reaction by which it does so goes faster in the presence of the molecules typically found on the surface of many bacteria and viruses.
Like the blood-clotting pathway, the complement pathway is a cascade. Inevitably, in both cases one encounters the same problems trying to imagine their gradual production. It is not the final activity of a cascade that is the problem. The formation of a hole in a membrane does not necessarily require several different components; one killer protein could conceivably do the job. Nor does the formation of a protein aggregate, such as in blood clotting, necessarily require multiple components; under the right conditions, any protein will aggregate. (The particular shapes of the complement hole-complex and fibrin aggregate, however, are particularly suited to the jobs they do and need to be explained.) And as we saw in Chapter 4, a telephone pole by itself could bop Foghorn Leghorn. It is the control systems that are the problem. At each control point both the regulatory protein and the masked protein that it activates have to be present from the beginning. If C5b were present, the rest of the cascade would immediately be touched off; but if C5 were present with nothing to activate it, then the whole pathway would always be shut off. If C3b were present, the rest of the cascade would immediately be touched off; but if C3 were present with nothing to activate it, then the whole pathway would always be shut off. Even if one imagines a much shortened pathway (where, say, Cls directly cuts C5), insertion of additional control points into the middle of the cascade runs into the same problem: the irreducible complexity of the switches.
In addition to the generic problems of setting up a cascade, the complement pathway shares another problem with the blood-clotting cascade: attachment of proteins to membranes is crucial. Several clotting factors must first be modified to synthesize Gla residues so that they could stick to a membrane. In the complement pathway, both C3 and C4 have unusual, highly reactive internal groups that chemically attach to the membrane after the proteins are cleaved by other factors. These special features have to be available before the pathway is functional, adding a further severe barrier to their gradual development. Numerous little features of the complement system are stumbling blocks to gradual development. Let's consider some subtle characteristics of just the C1 system. The three types of proteins in C1q braid around each other, but do not braid with themselves. If they did, then the ratio of different types of chains in the complex would be changed, and there would be a much smaller chance of getting the real C1q complex with six copies of three different chains. If the binding of C1q to the antibody-foreign cell did not trigger C1r's self-scission, then the cascade would be stopped in its tracks. Conversely, if C1r cut itself before C1q bound to the antibody complex, then the cascade would be prematurely triggered. And so on.
The proper functioning of the immune system is a prerequisite for health. Major illnesses such as cancer and AIDS have either their cause or their cure, or both, in the vagaries of the system. Because of its impact on public health, the immune system is a subject of intense interest. Thousands of research laboratories around the world work on various aspects of the immune system. Their efforts have already saved many lives and promise to save many more in the future.
Although great strides have been made in understanding how the immune system works, we remain ignorant of how it came to be. None of the questions raised has been answered by any of the thousands of scientists in the field; few have even asked the questions. A search of the immunological literature shows ongoing work in comparative immunology (the study of immune systems from various species). But that work, valuable though it is, does not address in molecular detail the question of how immune systems originated. Perhaps the best efforts at doing that so far have been in two short papers. The first, by Nobel laureate David Baltimore and two other prominent scientists, is tantalizingly entitled «Molecular Evolution of the Vertebrate Immune System.» But it's hard to live up to such a title in just two pages. The authors point out that for any organism to have an immune system akin to that seen in mammals, the minimally required molecules are the antigen receptors (immunoglobulin and TCR), the antigen presentation molecules (MHC), and the gene rearranging proteins. (Immunoglobulins are antibodies. TCR molecules are akin to antibodies.) The authors then argue that sharks, which are very distantly related to mammals, appear to have all three components. But it's one thing to say an organism has a completed, functioning system, and another to say how the system developed. The authors certainly realize this. They note that immunoglobulin and TCR genes both require RAG proteins for rearrangement. On the other hand, RAG proteins require specific recombination signals to rearrange immunoglobulin and TCR genes. (RAG is the component that rearranges the genes.) They make a valiant stab at accounting for the components, but in the end, it is a hop in the box with Calvin and Hobbes. The authors speculate that a gene from a bacterium might have luckily been transferred to an animal. Luckily, the protein coded by the gene could itself rearrange genes; and luckily, in the animal's DNA there were signals that were near antibody genes; and so on. In the final analysis the authors identify key problems with gradualistic evolution of the immune system, but their proffered solutions are really just a disguised shrug of their shoulders.
Another paper that gamely tries to account for a piece of the immune system is entitled «Evolution of the Complement System.»6 Like the paper discussed above, it is very short and is a commentary article—in other words, not a research article. The authors make some imaginative guesses about what might come first and second, but inevitably they join Russell Doolittle in proposing unexplained proteins that are «unleashed» and «spring forth» («At some point a critical gene fusion created a protease with a binding site for the primitive C3b»; «Evolution of the other alternative pathway components further improved the amplification and specificity»; and «C2, created by the duplication of the factor В gene, would then have allowed further divergence and specialization of the two pathways»). No quantitative calculations appear in the paper. Nor does an acknowledgment that gene duplications would not immediately make a new protein. Nor does any worry about a lack of controls to regulate the pathway. But then, it would be hard to fit those concerns in the four paragraphs of the paper that deal with molecular mechanisms.
There are other papers and books that discuss the evolution of the immune system.7 Most of them, however, are at the level of cell biology and thus unconcerned with detailed molecular mechanisms, or else they are concerned simply with comparison of DNA or protein sequences. Comparing sequences might be a good way to study relatedness, but the results can't tell us anything about the mechanism that first produced the systems.
We can look high or we can look low, in books or in journals, but the result is the same. The scientific literature has no answers to the question of the origin of the immune system.
I have looked at three features of the immune system—clonal selection, antibody diversity, and the complement system—and demonstrated that each individually poses massive challenges to a putative step-by-step evolution. But showing that the parts can't be built step by step only tells part of the story, because the parts interact with each other. Just as a car without steering, or a battery, or a carburetor isn't going to do you much good, an animal that has a clonal selection system won't get much benefit out of it if there is no way to generate antibody diversity. A large repertoire of antibodies won't do much good if there is no system to kill invaders. A system to kill invaders won't do much good if there's no way to identify them. At each step we are stopped not only by local system problems, but also by requirements of the integrated system.
clonal selection
antibody diversity
system to kill invaders
a way to identify them
Last edited by Admin on Tue Jan 29, 2019 1:59 pm; edited 3 times in total