Dr. John Sanford Lectures on Inevitable Genomic DeteriorationSean Pitman / February 20, 2012
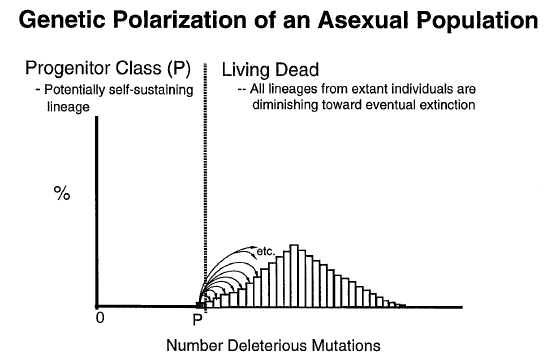
Dr. John C. Sanford, well-known scientist, Associate Professor of at Cornell University, plant geneticist, and inventor of the “gene gun”, recently gave a talk at Loma Linda University (as part of Dr. Paul Giem’s Creation lecture series) on “Genomic Entropy” or the inevitable genetic deterioration and eventual extinction of humans within a relatively short period of time (hence the title of his talk “Down, Not Up”). In other words, random detrimental mutations build up in the gene pools of living things with a low reproductive rate far far faster than natural selection can get rid of them. This generally accepted fact of modern science strongly implies, therefore, that we have devolved, not evolved, from an originally superior state, as a species or collective gene pool, compared to our current rapidly degenerating condition. We are all heading downhill, genetically, very rapidly in fact, toward complete genetic meltdown – toward the extinction of life on this planet from the build-up of deleterious mutations alone. The only solution to this problem is the direct intervention of the Creator of life. It is for this reason that Dr. Sanford likes to call himself a “Supernaturalist”.
Dr. Sandford’s current position as a believer in God as the original Creator of life was not always his position. He was once an atheist. He then evolved, if you will, into a theistic position, and then, finally, was convinced of the superior explanatory value of the creationist position – of a belief in the scientific credibility of the Biblical account of origins and the current degenerated and degenerating condition of mankind.
For those interested, here is my own presentation on the differences between genetic and thermodynamic entropy (often confused by both creationists and mainstream scientists) and the inevitable steady genetic decline in quality described by Dr. Sanford… a concept I like to call,
“Turtles All the Way Down”:
.
The following is a summary of the facts involved with Dr. Sanford’s conclusions. For instance, what is the evidence for the notion that the rate of accumulation of the detriment mutations outstrips any mechanisms for their removal from a given gene pool?
Well, the answer to this question is based on known overall mutation rates, the amount of functional DNA in the genome, the ratio of beneficial vs. detrimental mutations, and the known reproductive/death rate that would be required to compensate for the detrimental mutation rate.
Summary
Overall mutation rate: A fairly recent paper in a 2010 issue of
Science attempted a direct measurement of the mutation rate by comparing the complete genome sequences of two offspring and their parents. They estimate that each offspring had only 70 new mutations (instead of previously predicted rates of around 170) for an overall mutation rate of around 1.1 * 10
-8 per site per generation (Roach et al. 2010: Link). Another paper published in a 2010 issue of PNAS suggested an overall autosomal mutation rate of 1.481 * 10
−8 base substitutions per site per generation – or approximately 89 new mutations per person per generation (Lynch, 2009: Link). Unfortunately for men, a 2009 pedigree-based estimate derived from high-throughput sequencing of Y chromosomes (~58 million bp) separated by 13 generations (Xue et al. 2009: Link) yielded a much higher base-substitutional mutation rate estimate of 3.0 * 10
−8 for the Y-chromosome (~ 1.74 mutations per person, per Y-chromosome alone, per generation – – comparable to a rate of ~180 autosomal mutations per person per generation).
For purposes of discussion, let’s assume an average SNP mutation rate of 70 per person, per generation.
Comment:Note, however, that this mutation rate only represents
point mutations. A mutation rate of 70 isn’t truly representative of all types of mutations – such as deletions, insertions, duplications, translocations, inversions, micro-satellite mutations, various forms of indel mutations, etc. So, the actual mutation rate with regard to the absolute number of nucleotide changes over time would be higher than this. Consider, for example, that although “macro-mutations” (like larger insertions or deletions) occur at a rate of an additional 4-12 per person per generation, they actually change
100-500 times the number of nucleotides that are changed by all point mutations combined. So, the additional
effective nucleotide mutation rate could be up to
30,000nucleotide changes per person per generation. (Link).
Functional DNA in the Genome:
In the past five years or so, the discovery that a significant amount of “non-coding DNA” is functional to one degree or another. Early on, it was thought that no more than 1.5% of the human genome was functional. Although there are about 23,000 protein-coding genes, these comprise a mere 1.5% of the human genome. The rest of the genome is comprised of DNA sequences that do not code for proteins. It is interesting to note that about 80% of the non-coding DNA in the human genome is actually transcribed (Link), mainly into non-protein-coding RNAs (Link). Many of the observed non-coding transcripts are differentially expressed, and, while most have not yet been studied, increasing numbers are being shown to be functional and/or trafficked to specific subcellular locations, as well as exhibit subtle evidence of selection. Even some of the 20% or so of the genome that is not transcribed at all into any form of RNA, such as repetitive sequences, is being shown to have functionality (in regulation of gene expression, overall chromosome structure and pairing, etc). For example, the non-transcribed spacer (NTS) region of rRNA genes is the “most important region of the rDNA” because this is the region that contains the nucleotide sequences that trigger and/or terminate transcription (Link).
Of course, analyses of conservation patterns indicate that only 5% (3% – 8%) of the human genome is under purifying selection for functions common to mammals. However, these estimates rely on the assumption that reference sequences (usually sequences thought to be ancient transposon-derived sequences) have evolved neutrally, which may not be the case (especially if common descent theories are wrong), and if so would lead to an underestimate of the fraction of the genome under selective constraint. These analyses also do not detect functional sequences that are evolving rapidly and/or have acquired lineage-specific functions. Indeed, many regulatory sequences and known functional noncoding RNAs, including many microRNAs, are not conserved over significant evolutionary distances, and recent evidence from the ENCODE project suggests that many functional elements show no detectable level of sequence constraint. Also, a 2010 report on research by Kunarso et al. in
Nature suggests:
“Although sequence conservation has proven useful as a predictor of functional regulatory elements in the genome the observations by Kunarso
et. al. are a reminder that it is not justified to assume in turn that all functional regulatory elements show evidence of sequence constraint.” (Link)
Some even go on to argue that, “It is possible that much if not most of the human genome may be functional.” (Pheasant and attick, 2007: Link) From the conclusion of their paper, Pheasant and Mattick write:
“It seems clear that 5% is a minimum estimate of the fraction of the human genome that is functional, and that the true extent is likely to be significantly greater. If the upper figure of 11.8% under common purifying selection in mammals from ENCODE (Margulies et al. 2007) is realistic across the genome as a whole, and if turnover and positive selection approximately doubles this figure (Smith et al. 2004), then the functional portion of the genome may exceed 20%. It is also now clear that the majority of the mammalian genome is expressed and that many mammalian genes are accompanied by extensive regulatory regions. Thus, although admittedly on the basis of as yet limited evidence, it is quite plausible that many, if not the majority, of the expressed transcripts are functional and that a major component of genomic information is rapidly evolving regulatory DNA and RNA. Consequently, it is possible that much if not most of the human genome may be functional. This possibility cannot be ruled out on the available evidence, either from conservation analysis or from genetic studies (Mattick and Makunin 2006), but does challenge current conceptions of the extent of functionality of the human genome and the nature of the genetic programming of humans and other complex organisms.”
The science journal
Nature also published a very interesting news feature along these lines (ENCODE: The human encyclopaedia, Sept 5, 2012). This article reports on the ongoing human genome project called the “Encyclopedia of DNA Elements” or ENCODE project where the researchers assigned function to much of what was previously described as “junk DNA” – going so far as to suggest functionality of at least 80% of the human genome. While this suggestion is likely a bit extreme, an estimate of at least 20% functionality does seem fairly conservative at the present time (Kellis, 2014).
Implied functional mutation rate::
Given that 20% of the genome is functional to one degree or another, this would imply a functional (non-neutral) mutation rate of 11 per person per generation (70 total mutations times 20% times the number of non-redundant or non-synonymous mutations at about 80%). This is in line with the most conservative estimates recently published in literature. For example, Kellis (2014) argues that:
“The lower bound estimate that 5% of the human genome has been under evolutionary constraint was based on the excess conservation observed in mammalian alignments relative to a neutral reference (typically ancestral repeats, small introns, or fourfold degenerate codon positions). However, estimates that incorporate alternate references, shape-based constraint, evolutionary turnover, or lineage-specific constraint each suggests roughly two to three times more constraint than previously (12-15%), and their union might be even larger as they each correct different aspects of alignment-based excess constraint…. Although still weakly powered, human population studies suggest that an additional 4-11% of the genome may be under lineage-specific constraint after specifically excluding protein-coding regions.”
This means that, at minimum, between 16% to 26% of the genome is likely to be functionally constrained to one degree or another. And, of course, this means that the likely detrimental mutation rate is at least four times as high as Keightley suggested in 2012 (and some would argue even higher) – i.e., about 8.8 detrimental mutations per offspring per generation. This would, of course, imply a necessary reproductive rate of over 13,200 offspring per woman per generation (and a death rate of over 99.99% per generation).
Ratio of beneficial vs. detrimental mutations:
There are numerous published estimates ranging from 1/1000 to 1/1,000,000. A 1998 paper published in Genetica suggests a beneficial mutation rate (vs. the total mutation rate) of approximately 1 in 1,000,000 (Gerrish and Lenski, 1998: [url=http://myxo.css.msu.edu/lenski/pdf/1998, Genetica, Gerrish & Lenski.pdf]Link[/url]). Given that a significant portion if not most of the human genome is functional to one degree or another, to a similar degree those mutations that are not beneficial would be functionally detrimental to one degree or another. In short, the ratio of beneficial vs. detrimental is very small – most likely well below the ratio of 1/1000.
Detrimental mutation rate:
Given that the ratio of beneficial vs. detrimental mutations is so low (less than 1/1000), the detrimental mutation rate would be very similar to the overall functional mutation rate. In other words there would be between around 11 detrimental mutations (to include mostly near-neutral detrimental mutations) per person per generation (with a more conservative estimate of at least 8 detrimental mutations; see discussion above).
Required reproductive/death rate to compensate for detrimental mutation rate:
The reduction in fitness (i.e., the genetic load) due to deleterious mutations with multiplicative effects is given by the formula of 1 – e
-U (Kimura and Moruyama, 1966). For a detrimental mutation rate (U
d) of just 3 mutations per person per generation, the average fitness is reduced to 1 – 2.71828183
-3 = 0.95 of the original parental fitness level. The number of offspring, in a sexually reproducing species, needed to maintain the population at the parental level of fitness would therefore be: 1 / e
-3 = 20 offspring per woman per generation for just one to survive without any detrimental mutations. Therefore, each woman would need to produce 40 offspring for 2 to survive without any detrimental mutations to maintain the population at functional genetic neutrality (at least a 90% death rate without considering genetically non-related accidents). Of course, if the detrimental mutation rate were really more like 11 per person per generation, the number of offspring needed, per woman, to allow natural selection to deal with this degree of bad karma would be around 2 * 1/e
-11= ~120,000 offspring per woman per generation. Even with a much more conservative estimate of U = 8, the required reproductive rate would be about 6,000 per woman per generation (quite clearly an impossibility either way).
Now, one might argue that the actual detrimental mutation rate is much lower than this, but it is rather hard to believe that the minimum number of offspring required per woman would be remotely within the realm of feasibility, given what we’ve learned about the functionality of the non-coding elements of the genome in recent years. Humans simply do not reproduce remotely fast enough to keep up with the most conservative understanding of the minimum rate of detrimental mutations that hits every single member of the human gene pool in every generation.
Consider also that Hermann Joseph Muller, a famous pioneer in the field of genetics, argued that a detrimental mutation rate of just 0.5/person/generation (an average reproductive rate of 3 children per woman) would doom the human population to eventual extinction (H. J. Muller, 1950). After all, it was Muller who realized that, in effect, each detrimental mutation leads, ultimately, to one “genetic death,” since each mutation can be eliminated only by death or failure to reproduce. Sexual recombination softens this conclusion somewhat (by about half), but does not really solve the problem – as discussed above. Also, various forms of truncation selection and quasi-truncation selection (Link) and positive epistasis (discussed above) really don’t solve a problem of this magnitude either.
Within mainstream literature clear limitations to mutation rates are known because of this particular problem. Even rapidly reproducing bacteria and viruses have a fairly small limit to the number of mutations that can be sustained per generation. Based on research coming out of Harvard University, that number is less than 6 mutations per individual per generation – for bacteria and viruses as well as most other living things! This is a total number of mutations affecting functional regions of DNA – counting detrimental, beneficial, and neutral varieties.
“If enough mutations push an essential protein towards an unstable, non-functional structure, the organism will die. Shakhnovich’s group found that for most organisms, including viruses and bacteria, an organism’s rate of genome mutation must stay below 6 mutations per genome per generation to prevent the accumulation of too many potentially lethal changes in genetic material.” (Link, Link-2, Link-3)
For viruses in particular, the limiting mutation rate was found to be just 2.5 mutations per genome per generation (Link). This is the total mutation rate, not just the detrimental mutation rate. Also, the population here is assumed to be infinite in size. For finite populations the maximum tolerable mutation rate would obviously be smaller. The smaller the population, the lower the mutation rate that can be tolerated without an eventual genetic meltdown.
But what about the effect of beneficial mutations?
“Whitlock included beneficial mutations and calculated that N
crit ~(U
deleterious/U
beneficial)
1/3, which depends only on the balance of beneficial to deleterious mutations and not on the mutation rate itself. Both of those examples contradict our results, which show that N
crit and τ depend dramatically on |U|. The dominant reason for the discrepancy is that those authors assumed that deleterious mutations occur ‘one at a time,’ which is not true when the rate that mutations are introduced (U) exceeds the rate at which selection removes them (~1/s). When U/s>>1, the population experiences ‘Hill-Robertson interference’, which both accelerates extinction and also makes analytic solutions intractable.” (Link)
The Y-Chromosome Rapidly Headed for Extinction?
Also, what about the Y-chromosome in males? The Y-chromosome does not undergo significant sexual recombination. Are the males of slowly reproducing species, like humans, therefore headed for extinction at an even faster rate than females?
“The absence of recombination with a homologous partner means that it [The Y-chromosome] can never be repaired by recombination. This has led to suggestions that the Y is destined for extinction it will eventually dwindle to nothing. According to this model, its role in sex determination will eventually be taken on by genes elsewhere in the genome.”
50The author of the above quoted article goes onto point out that several species, like the Armenian mole vole, are able to reproduce without the Y chromosome. While this might explain where humans are headed, it doesn’t seem quite clear as to just how the Y-chromosome could have evolved over millions of years of time given its relative inability to combat high detrimental mutation rates in humans. Of course, research (Jennifer Hughes, 2012) has shown that the human Y-chromosome is “remarkably similar” to that of the rhesus monkey. In an interview Hughes noted that, “For the most part, the gene content [for human vs. rhesus Y-chromosomes] has not changed for 25 million years.” (Link). Based on the assumed evolutionary relationship between humans and monkeys, it seems then like men are not going extinct after all! – Phew! However, when one considers how high the actual detrimental mutation rate of the Y-chromosome really is (Link), it not only calls into serious question the survival of the Y-chromosome, but the entire notion of human-ape common descent from a shared common ancestor. In fact, this conclusion seems much more consistent with a study published a bit earlier in
Nature (Hughes, 2010) that showed many striking differences between human and chimp chromosome structure, gene content, and even qualitatively unique genes between the two species. As far as looking at specific genes, the chimp and human Y-chromosomes seem to have a dramatic difference in gene content of up to 53%. In other words, the chimp is lacking approximately half of the genes found on a human Y-chromosome. Because genes occur in families or similarity categories, the researchers also sought to determine if there was any difference in actual gene categories. They found a shocking 33 percent difference. The human Y-chromosome contains a third more gene categories, entirely different
classes of genes, compared to chimps.
Under evolutionary assumptions of long and gradual genetic changes, the Y-chromosome structures, layouts, genes, and other sequences should be much the same in both species, given only six million years or so since chimpanzees and humans supposedly diverged from a common ancestor. Instead, the differences between the Y-chromosomes are marked. R. Scott Hawley, a genetics researcher at the Stowers Institute in Kansas City, though not involved in the research, told the Associated Press, “That result is astounding.” (Link)
Because virtually every structural aspect of the human and chimp Y-chromosomes is different, it is hard to arrive at an overall similarity estimate between the two. The researchers did postulate an overall 70 percent similarity, which did not take into account size differences or structural arrangement differences. This was done by concluding that only 70 percent of the chimp sequence could be aligned with the human sequence – not taking into account differences within the alignments.
In other words, 70 percent was a conservative estimate, especially when considering that 50 percent of the human genes were missing from the chimp, and that the regions that did have some similarity were located in completely different patterns. When all aspects of non-similarity (sequence categories, genes, gene families, and gene position) are taken into account, it is safe to say that the overall similarity is actually much lower than 70 percent. In fact, this difference is so striking that the authors of the
Nature article described the discrepancy with the standard evolutionary model in a rather intriguing way:
“Indeed, at 6 million years of separation, the difference in MSY gene content in chimpanzee and human is more comparable to the difference in autosomal gene content in chicken and human, at 310 million years of separation.” (Link)
Given the standard evolutionary model of origins, it is indeed rather stunning to consider that the human Y-chromosome looks just as different from a chimp as the other human chromosomes do from a chicken. How is this explained within the evolutionary mindset? Obviously, the believer in mainstream evolutionary models is now forced to invent more just-so stories of major chromosomal rearrangements and rapid generation of many new genes, along with vast amounts of regulatory DNA, within very short spans of evolutionary time. However, since each respective Y-chromosome appears fully integrated and interdependently stable with its host organism, the most logical inference from the Y-chromosome data, without any prior commitment to the evolutionary story of origins, is that humans and chimpanzees were each specially created as distinct creatures, or evolved over a far far greater period of time…
Additional research carried out in 2012 by scientists at the University of Oxford and the University of Chicago found that hotspot regions that determine the locations for genetic recombination during cellular meiosis in sexual reproduction showed “no overlap between humans and chimpanzees.” (Link) This was an “extraordinarily unexpected finding” given the other similarities between humans and chimps. Professor McVean explains:
“Genetic recombination has been likened to shuffling a deck of cards, which ensures that children are given a different genetic ‘hand’ than their parents. We know that in many cases recombination occurs where a particular thirteen letter sequence is present — this is like a run of hearts from ace to king determining where we cut the deck of cards. Because humans and chimpanzees are genetically very similar, we might explain that you can only ‘cut the cards’ at the same point — in fact, we find that this is not true.” (Link)
Additional research carried out in 2012 by scientists at the University of Oxford and the University of Chicago found that hotspot regions that determine the locations for genetic recombination during cellular meiosis in sexual reproduction showed “no overlap between humans and chimpanzees.” (Link) This was an “extraordinarily unexpected finding” given the other similarities between humans and chimps. Professor McVean explains:
“Genetic recombination has been likened to shuffling a deck of cards, which ensures that children are given a different genetic ‘hand’ than their parents. We know that in many cases recombination occurs where a particular thirteen letter sequence is present — this is like a run of hearts from ace to king determining where we cut the deck of cards. Because humans and chimpanzees are genetically very similar, we might explain that you can only ‘cut the cards’ at the same point — in fact, we find that this is not true.” (Link)
http://www.educatetruth.com/featured/dr-john-sanford-lectures-on-inevitable-genomic-deterioration/





 "If enough mutations push an essential protein towards an unstable, non-functional structure, the organism will die. Shakhnovich's group found that for most organisms, including viruses and bacteria, an organism's rate of genome mutation must stay below 6 mutations per genome per generation to prevent the accumulation of too many potentially lethal changes in genetic material." (Link, Link-2, Link-3)
"If enough mutations push an essential protein towards an unstable, non-functional structure, the organism will die. Shakhnovich's group found that for most organisms, including viruses and bacteria, an organism's rate of genome mutation must stay below 6 mutations per genome per generation to prevent the accumulation of too many potentially lethal changes in genetic material." (Link, Link-2, Link-3)