The awe inspiring spliceosome, the most complex macromolecular machine known, and pre-mRNA processing in eukaryotic cells
https://reasonandscience.catsboard.com/t2180-the-spliceosome-the-splicing-code-and-pre-mrna-processing-in-eukaryotic-cells
Along the way to make proteins in eukaryotic cells, there is a whole chain of subsequent events that must all be fully operational, and the machinery in place, in order to get the functional product, that is proteins. At the beginning of the process, DNA is transcribed in the RNA polymerase molecular machine, to yield messenger RNA ( mRNA ), which afterward must go through post-transcriptional modifications. That is CAPPING, ELONGATION, SPLICING, CLEAVAGE, POLYADENYLATION, AND TERMINATION before it can be EXPORTED FROM THE NUCLEUS TO THE CYTOSOL, and PROTEIN SYNTHESIS INITIATED, (TRANSLATION), and COMPLETION OF PROTEIN SYNTHESIS AND PROTEIN FOLDING.
Bacterial mRNAs are synthesized by the RNA polymerase starting and stopping at specific spots on the genome. The situation in eukaryotes is substantially different. In particular, transcription is only the first of several steps needed to produce a mature mRNA molecule. The mature transcript for many genes is encoded in a discontinuous manner in a series of discrete exons, which are separated from each other along the DNA strand by non-coding introns. mRNAs, rRNAs, and tRNAs can all contain introns that must be removed from precursor RNAs to produce functional molecules.The formidable task of identifying and splicing together exons among all the intronic RNA is performed by a large ribonucleoprotein machine, the spliceosome, which is composed of several individual small nuclear ribonucleoproteins, five snRNPs, pronounced ‘snurps’, (U1, U2, U4, U5, and U6) each containing an RNA molecule called an snRNA that is usually 100–300 nucleotides long, plus additional protein factors that recognize specific sequences in the mRNA or promote conformational rearrangements in the spliceosome required for the splicing reaction to progress, and many more additional proteins that come and go during the splicing reaction. It has been described as one of "the most complex macromolecular machines known," "composed of as many as 300 distinct proteins and five RNAs".
The snRNAs perform many of the spliceosome’s mRNA recognition events. Splice site consensus sequences are recognized by non-snRNP factors; the branch-point sequence is recognized by the branch-point-binding protein (BBP), and the polypyrimidine tract and 3′ splice site are bound by two specific protein components of a splicing complex referred to as U2AF (U2 auxiliary factor), U2AF65 and U2AF35, respectively.
This is one more great example of an amazingly complex molecular machine, that will operate and exercise its precise orchestrated function properly ONLY with ALL components fully developed and formed and able to interact in a highly complex, ordered precise manner. Both, the software, and the hardware must be in place fully developed, or the machine will not work. No intermediate stage will do the job. And neither would snRNPs (U1, U2, U4, U5, and U6) have any function if not fully developed. And even if they were there, without the branch-point-binding protein (BBP) in place, nothing done, either, since the correct splice site could not be recognized. Had the introns and exons not have to emerge simultaneously with the Spliceosome? No wonder, does the paper: " Origin and evolution of spliceosomal introns " admit: Evolution of the exon-intron structure of eukaryotic genes has been a matter of long-standing, intensive debate. 1 and it concludes that: The elucidation of the general scenario of the evolution of eukaryote gene architecture by no account implies that the main problems in the study of intron evolution and function have been solved. Quite the contrary, fundamental questions remain wide open. If the first evolutionary step would have been the arise of self-splicing Group II introns, then the question would follow: Why would evolution not have stopped there, since that method works just fine?
There is no credible road map, how introns and exons, and the splice function could have emerged gradually. What good would the spliceosome be good for, if the essential sequence elements to recognize where to slice would not be in place? What would happen, if the pre mRNA with exons and introns were in place, but no spliceosome ready in place to do the post-transcriptional modification, and neither the splicing code, which directs the way where to splice? In the article: ‘JUNK’ DNA HIDES ASSEMBLY INSTRUCTIONS, the author, Wang, observes that splicing "is a tightly regulated process, and a great number of diseases are caused by the 'misregulation' of splicing in which the gene was not cut and pasted correctly." Missplicing in the cell can have dire consequences as the desired product is not produced, and often the wrong products can be toxic for the cell. For this reason, it has been proposed that ATPases are important for ‘proofreading’ mechanisms that promote fidelity in splice site selection.
In his textbook Essentials of Molecular Biology, George Malacinski points out why proper polypeptide production is critical:
"A cell cannot, of course, afford to miss any of the splice junctions by even a single nucleotide, because this could result in an interruption of the correct reading frame, leading to a truncated protein."
The required precision is quite amazing, and even more astounding is the fact that these incredibly complex molecular machines are able and capable to do the job in the precise manner as needed.
Following the binding of these initial components, the remainder of the splicing apparatus assembles around them, in some cases displacing some of the previously bound components.
Question: How could the information to assemble the splicing apparatus correctly have emerged gradually? In order to do so, had the assembly parts not have to be there, at the assembly site, fully developed, and ready for recruitment? Had the availability of these parts not have to be synchronized so that at some point, either individually or in combination, they were all available at the same time? Had the assembly not have to be coordinated in the right way right from the start? Had the parts not have to be mutually compatible, that is, ‘well-matched’ and capable of properly ‘interacting’? even if subsystems or parts are put together in the right order, they also need to interface correctly.
Is it feasible that this complex machine was the result of progressive evolutionary development, in which simple molecules are the start of the biosynthesis chain and are then progressively developed in sequential steps if the end goal is not known by the process and mechanism promoting the development? How could each intermediate in the pathway be an endpoint in the pathway, if that endpoint had no function? Did not each intermediate have to be usable in the past as an end product? And how could the be usable, if the amino acid sequence chain had only a fraction of the fully developed sequence? How could successive steps be added to improve the efficiency of a product where there was no use for it at this stage? Despite the fact that proponents of naturalism embrace this kind of scenario, it seems obvious that is extremely unlikely to be possible that way.
Martin and Koonin admit in their paper “Hypothesis: Introns and the origin of nucleus-cytosol compartmentalization,”: The transition to spliceosome-dependent splicing will also impose an unforgiving demand for inventions in addition to the spliceosome. And furthermore: More recent are the insights that there is virtually no evolutionary grade detectable in the origin of the spliceosome, which apparently was present in its (almost) fully-fledged state in the common ancestor of eukaryotic lineages studied so far. That's a surprising admittance.
This means that the spliceosome appeared fully formed almost abruptly and that the intron invasion took place over a short time and has not changed for supposedly hundreds of millions of years.
In another interesting paper : Breaking the second genetic code, the authors write 2 : The genetic instructions of complex organisms exhibit a counter-intuitive feature not shared by simpler genomes: nucleotide sequences coding for a protein (exons) are interrupted by other nucleotide regions that seem to hold no information (introns). This bizarre organization of genetic messages forces cells to remove introns from the precursor mRNA (pre-mRNA) and then splice together the exons to generate translatable instructions. An advantage of this mechanism is that it allows different cells to choose alternative means of pre-mRNA splicing and thus generates diverse messages from a single gene. The variant mRNAs can then encode different proteins
with distinct functions. One difficulty with understanding alternative pre-mRNA splicing is that the selection of particular exons in mature mRNAs is determined not only by intron sequences adjacent to the exon boundaries but also by a multitude of other sequence elements present in both exons and introns. These auxiliary sequences are recognized by regulatory factors that assist or prevent the function of the spliceosome — the molecular machinery in charge of intron removal.
Moreover, the coupling between RNA processing and gene transcription influences alternative splicing, and recent data implicate the packing of DNA with histone proteins and histone covalent modifications — the epigenetic code — in the regulation of splicing. The interplay between the histone and the splicing codes will, therefore, need to be accurately formulated in future approaches.
Question: How could natural mechanisms have provided the tuning, synchronization, and coordination between the histone and the splicing codes? First, these two codes and the carrier proteins and molecules ( the hardware and software ) would have to emerge by themselves, and in a second step orchestrate their coordination. Why is it reasonable to believe, that unguided, random chemical reactions would be capable of emerging with the immensely complex organismal functions?
Fazale Rana puts it nicely: Astounding is the fact that other codes, such as the histone binding code, transcription factor binding code, the splicing code, and the RNA secondary structure code, overlap the genetic code. Each of these codes plays a special role in gene expression, but they also must work together in a coherent integrated fashion.
1) http://www.biologydirect.com/content/7/1/11
2) http://nar.oxfordjournals.org/content/early/2013/11/07/nar.gkt1053.full.pdf
The spliceosome, the splicing code, and pre - mRNA processing in eukaryotic cells
Transcription Elongation in Eukaryotes Is Tightly Coupled to RNA Processing
Bacterial mRNAs are synthesized by the RNA polymerase starting and stopping at specific spots on the genome. The situation in eukaryotes is substantially different. In particular, transcription is only the first of several steps needed to produce a mature mRNA molecule. Other critical steps are the covalent modification of the ends of the RNA and the removal of intron sequences that are discarded from the middle of the RNA transcript by the process of RNA splicing
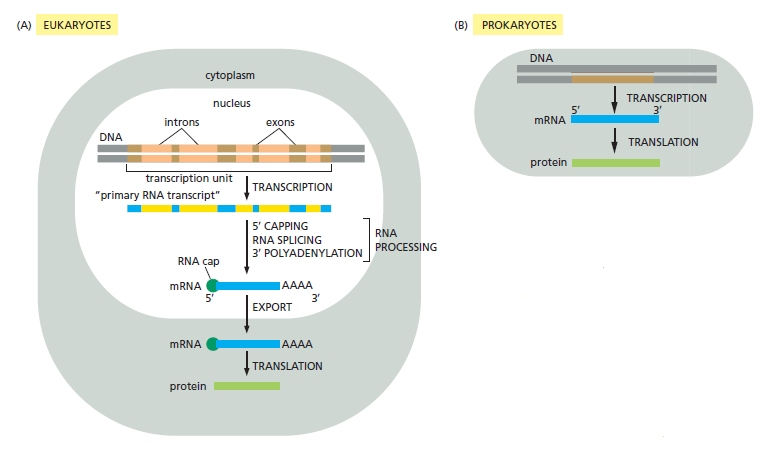
Comparison of the steps leading from gene to protein in eukaryotes and bacteria. The final level of a protein in the cell depends on the efficiency of each step and on the rates of degradation of the RNA and protein molecules.
(A) In eukaryotic cells, the mRNA molecule resulting from transcription contains both coding (exon) and noncoding (intron) sequences. Before it can be translated into protein, the two ends of the RNA are modified, the introns are removed by an enzymatically catalyzed RNA splicing reaction, and the resulting mRNA is transported from the nucleus to the cytoplasm. For convenience, the steps in this figure are depicted as occurring one at a time; in reality, many occur concurrently. For example, the RNA cap is added and splicing begins before transcription has been completed. Because of the coupling between transcription and RNA processing,
intact primary transcripts—the full-length RNAs that would, in theory, be produced if no processing had occurred—are found only rarely.
(B) In prokaryotes, the production of mRNA is much simpler. The 5ʹ end of an mRNA molecule is produced by the initiation of transcription, and the 3ʹ end is produced by the termination of transcription. Since prokaryotic cells lack a nucleus, transcription and translation take place in a common compartment, and the translation of a bacterial mRNA often begins before its synthesis has been completed.
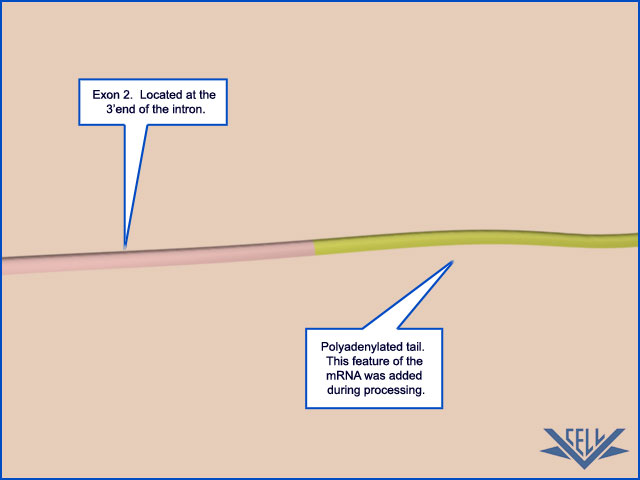
Both ends of eukaryotic mRNAs are modified: by capping on the 5ʹ end and by polyadenylation of the 3ʹ end 1 These special ends allow the cell to assess whether both ends of a mRNA molecule are present (and if the message is therefore intact) before it exports the RNA from the nucleus and translates it into protein.
Question: How could and would unguided, natural processes, where no intelligence is involved, emerge with that check out if both ends of mRNA are present? Quality control is usually an exercise attributed to intelligence.
RNA splicing joins together the different portions of a protein-coding sequence, and it provides eukaryotes with the ability to synthesize several different proteins from the same gene. A simple strategyhas evolved
Why not designed? figuring out what strategy to apply to get the best results is a mental process
to couple all of the above RNA processing steps to transcription elongation. As discussed previously, a key step in transcription initiation by RNA polymerase II is the phosphorylation of the RNA polymerase II tail, also called the CTD (C-terminal domain). This phosphorylation, which proceeds gradually as the RNA polymerase initiates transcription and moves along the DNA, not only helps dissociate the RNA polymerase II from other proteins present at the start point of transcription, but also allows a new set of proteins to associate with the RNA polymerase tail that function in transcription elongation and RNA processing. As discussed next, some of these processing proteins are thought to “hop” from the polymerase tail onto the nascent RNA molecule to begin processing it as it emerges from the RNA polymerase. Thus, we can view RNA polymerase II in its elongation mode as an RNA factory that not only moves along the DNA synthesizing an RNA molecule but also processes the RNA that it produces
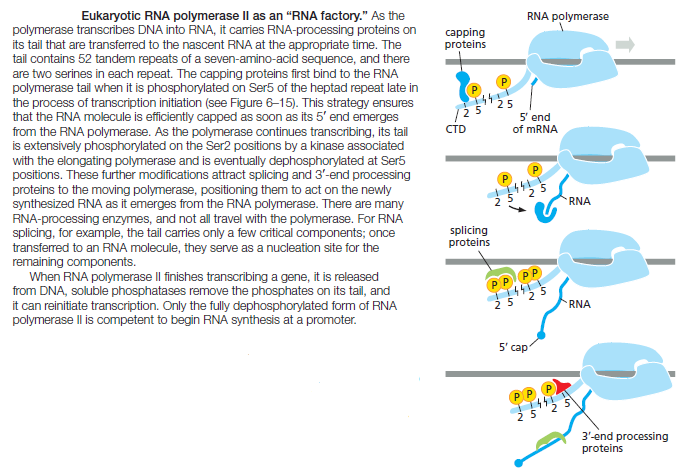
Fully extended, the CTD is nearly 10 times longer than the remainder of RNA polymerase. As a flexible protein domain, it serves as a scaffold or tether, holding a variety of proteins close by so that they can rapidly act when needed. This strategy, which greatly speeds up the overall rate of a series of consecutive reactions, is one that is commonly utilized in the cell.
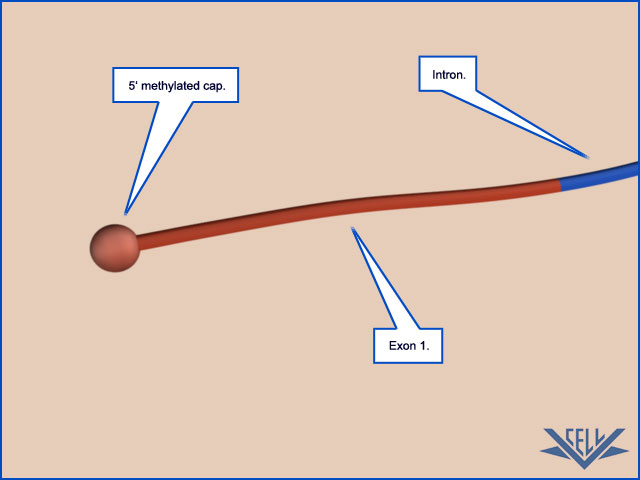
RNA Capping Is the First Modification of Eukaryotic Pre-mRNAs
As soon as RNA polymerase II has produced about 25 nucleotides of RNA, the 5ʹ end of the new RNA molecule is modified by addition of a cap that consists of a modified guanine nucleotide. Three enzymes, acting in succession, perform the capping reaction: one (a phosphatase) removes a phosphate from the 5ʹ end of the nascent RNA, another (a guanyl transferase) adds a GMP in a reverse linkage (5ʹ to 5ʹ instead of 5ʹ to 3ʹ), and a third (a methyl transferase) adds a methyl group to the guanosine
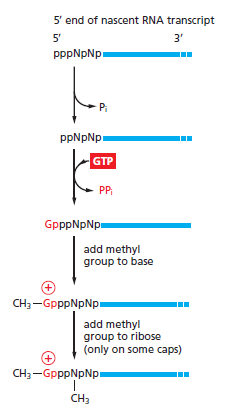
The reactions that cap the 5ʹ end of each RNA molecule synthesized by RNA polymerase II. The final cap contains a novel 5ʹ-to-5ʹ linkage between the positively charged 7-methyl G residue and the 5ʹ end of the RNA transcript . The letter N represents any one of the four ribonucleotides, although the nucleotide that starts an RNA chain is usually a purine (an A or a G).
Because all three enzymes bind to the RNA polymerase tail phosphorylated at the Ser5 position—the modification added by TFIIH during transcription initiation— they are poised to modify the 5ʹ end of the nascent transcript as soon as it emerges from the polymerase. The 5ʹ-methyl cap signifies the 5ʹ end of eukaryotic mRNAs, and this landmark helps the cell to distinguish mRNAs from the other types of RNA molecules present in the cell. For example, RNA polymerases I and III produce uncapped RNAs during transcription, in part because these polymerases lack a CTD. In the nucleus, the cap binds a protein complex called CBC (cap-binding complex), which, as we discuss in subsequent sections, helps a future mRNA be further processed and exported. The 5ʹ-methyl cap also has an important role in the translation of mRNAs in the cytosol, as we discuss later in the chapter.
RNA Splicing Removes Intron Sequences from Newly Transcribed Pre-mRNAs
The protein-coding sequences of eukaryotic genes are typically interrupted by noncoding intervening sequences (introns). Discovered in 1977, this feature of eukaryotic genes came as a surprise to scientists, who had been, until that time, familiar only with bacterial genes, which typically consist of a continuous stretch of coding DNA that is directly transcribed into mRNA. In marked contrast, eukaryotic genes were found to be broken up into small pieces of coding sequence (expressed sequences or exons) interspersed with much longer intervening sequences or introns; thus, the coding portion of a eukaryotic gene is often only a small fraction of the length of the gene
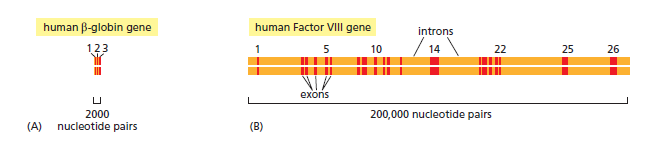
Structure of two human genes showing the arrangement of exons and introns.
(A) The relatively small β-globin gene, which encodes a subunit of the oxygen-carrying protein hemoglobin, contains 3 exons .
(B) The much larger Factor VIII gene contains 26 exons; it codes for a protein (Factor VIII) that functions in the bloodclotting pathway. The most prevalent form of hemophilia results from mutations in this gene.
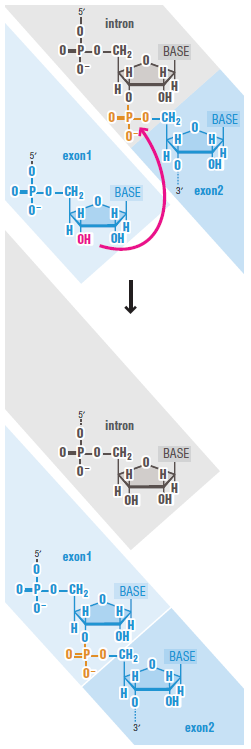
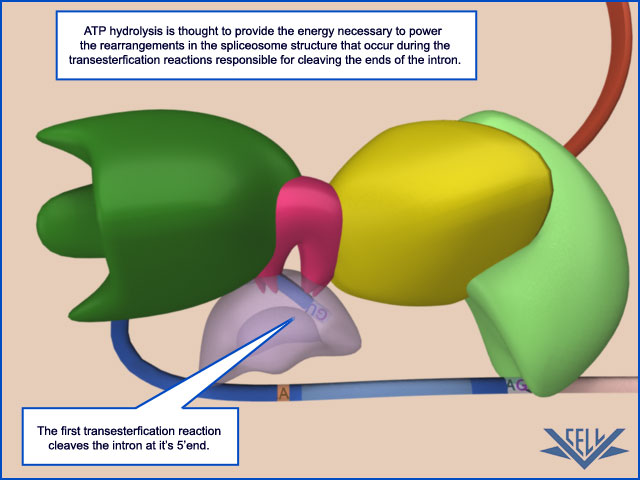
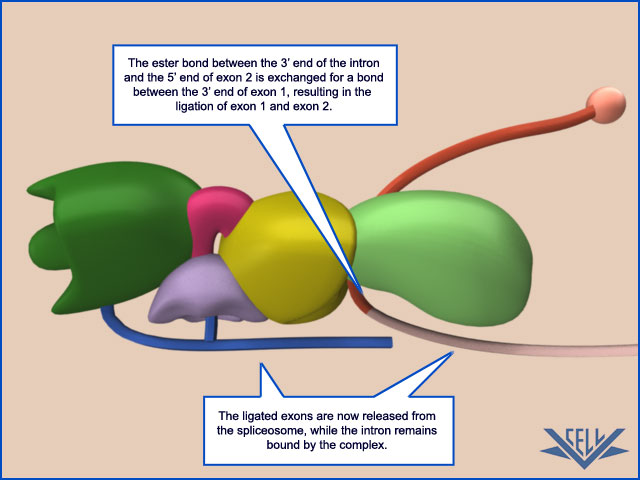
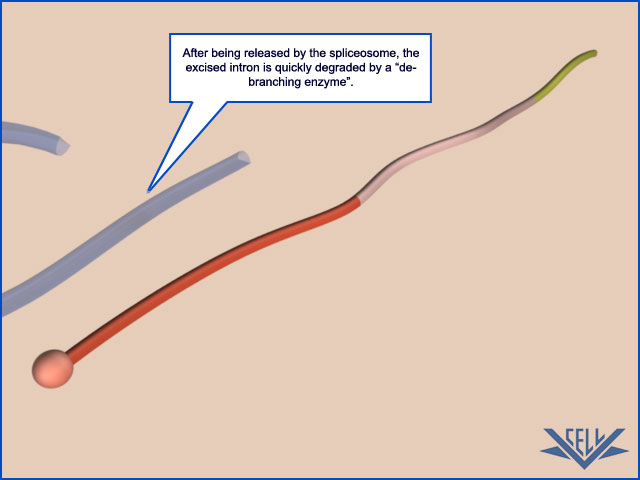
Both intron and exon sequences are transcribed into RNA. The intron sequences are removed from the newly synthesized RNA through the process of RNA splicing. The vast majority of RNA splicing that takes place in cells functions in the production of mRNA, and our discussion of splicing focuses on this so-called precursor-mRNA (or pre-mRNA) splicing. Only after 5ʹ- and 3ʹ-end processing and splicing have taken place is such RNA termed mRNA. Each splicing event removes one intron, proceeding through two sequential
phosphoryl-transfer reactions known as transesterifications; these join two exons together while removing the intron between them as a “lariat”

This is a highly ordered, sequential, and precise mechanism. How did this function emerge naturally? How was the specific adenine nucleotide put into the right place to be spliced? trial and error? In order to propose a natural origin, these questions need to be explained in a compelling manner.
The machinery that catalyzes pre-mRNA splicing is complex, consisting of five additional RNA molecules and several hundred proteins, and it hydrolyzes many ATP molecules per splicing event. This complexity ensures that splicing is accurate, while at the same time being flexible enough to deal with the enormous variety of introns found in a typical eukaryotic cell. It may seem wasteful to remove large numbers of introns by RNA splicing. In attempting to explain why it occurs, scientists have pointed out that the exon-intron arrangement would seem to facilitate the emergence of new and useful proteins over evolutionary timescales.
First of all, proponents of evolution should be able to explain why such a piece of complex machinery would emerge AT ALL, and why there should be a need of emergence of such complexity if bacterial cells work just fine without it. What I see here, is typical simplistic assertions without giving a considerable thought about the viability of random mutations to produce such enormous complexity
Thus, the presence of numerous introns in DNA allows genetic recombination to readily combine the exons of different genes, enabling genes for new proteins toevolve emerge more easily by the combination of parts of preexisting genes. RNA splicing also has a present-day advantage. The transcripts of many eukaryotic genes (estimated at 95% of genes in humans) are spliced in more than one way, thereby allowing the same gene to produce a corresponding set of different proteins.
Why should nature give preference to such a completely new method of producing a variety of proteins, rather than just let mutations and natural selection doing the job? It seems to me, in order to keep the pre-established fact of evolution, their proponents simply shut up with their critical thinking, and accept the fact of evolution, no matter how unlikely it seems
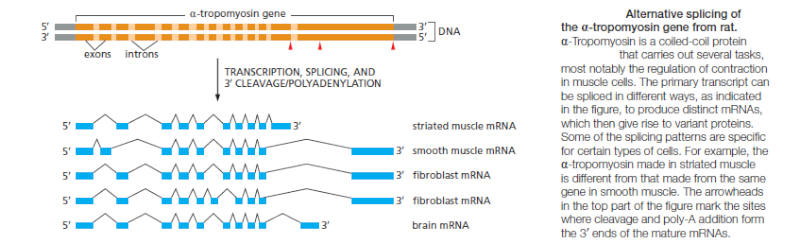
Rather than being the wasteful process it may have seemed at first sight, RNA splicing enables eukaryotes to increase the coding potential of their genomes.
In order to splice out the introns from exons, the cell required epigenetic information beside the complex machinery in place to do this complex task. Where did this information come from? trial and error?
Nucleotide Sequences Signal Where Splicing Occurs
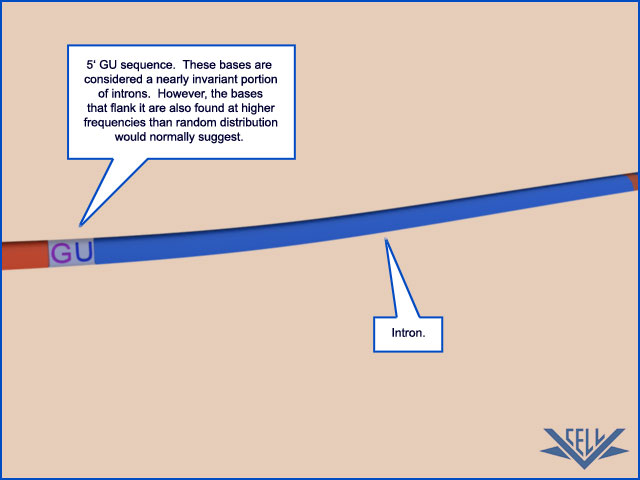
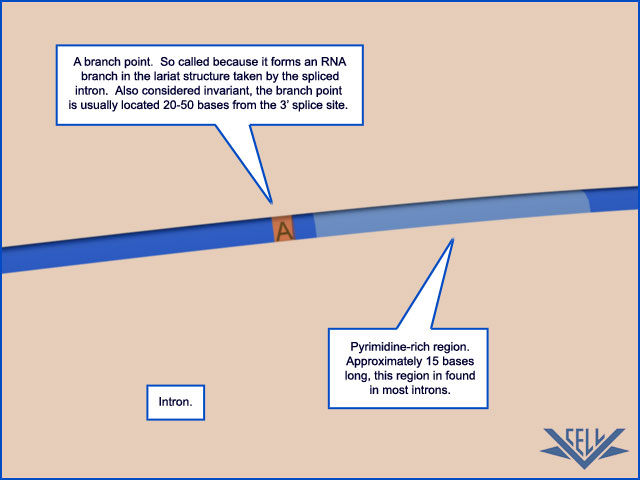
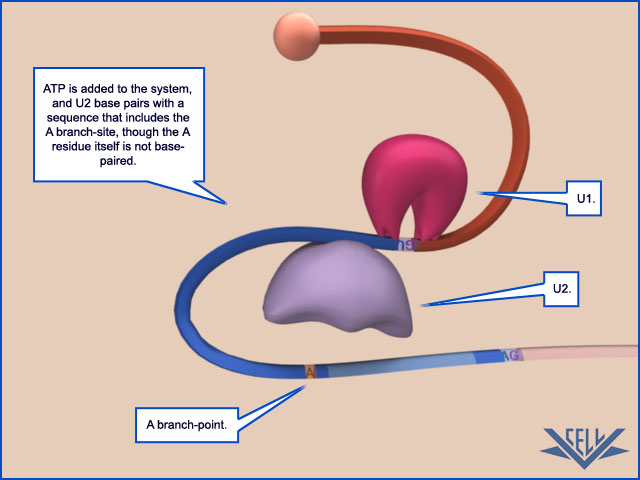
The mechanism of pre-mRNA splicing requires that the splicing machinery recognize three portions of the precursor RNA molecule: the 5ʹ splice site, the 3ʹ splice site, and the branch point in the intron sequence that forms the base of the excised lariat. Not surprisingly, each site has a consensus nucleotide sequence that is similar from intron to intron and provides the cell with cues for where splicing is to take place.
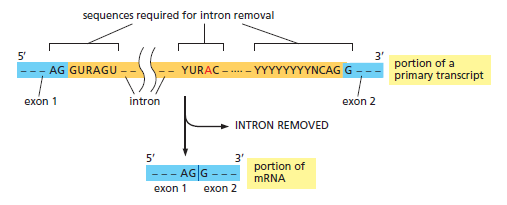
The consensus nucleotide sequences in an RNA molecule that signal the beginning and the end of most introns in humans. The three blocks of nucleotide sequences shown are required to remove an intron sequence. Here A, G, U, and C are the standard RNA nucleotides; R stands for purines (A or G); and Y stands for pyrimidines (C or U). The A highlighted in red forms the branch point of the lariat produced by splicing. Only the GU at the start of the intron and the AG at its end are invariant nucleotides in the splicing consensus sequences. Several different nucleotides can occupy the remaining positions, although the indicated nucleotides are preferred. The distances along the RNA between the three splicing consensus sequences are highly variable; however, the distance between the branch point and 3ʹ splice junction is typically much shorter than that between the 5ʹ splice junction and the branch point.
However, these consensus sequences are relatively short and can accommodate extensive sequence variability; the cell incorporates additional types of information to ultimately choose exactly where, on each RNA molecule, splicing is to take place.
Its evident that foresight is required a) to make the splicing machinery, and b) to program the information right from the start, where to slice; the whole mechanism has to arise all at once, fully functional. A stepwise arise is extremely unlikely, not to say, impossible. What good would the spliceosome be good for, if there were no information at the same time in order to make the machinery do its job? The hardware and the software are interdependent.
The high variability of the splicing consensus sequences presents a special challenge for scientists attempting to decipher genome sequences. Introns range in size from about 10 nucleotides to over 100,000 nucleotides, and choosing the precise borders of each intron is a difficult task even with the aid of powerful computers.
But cells " know " where to splice for a long time..... how did they " learn " that feat?
The possibility of alternative splicing compounds the problem of predicting protein sequences solely from a genome sequence. This difficulty is one of the main barriers to identifying all of the genes in a complete genome sequence, and it is one of the primary reasons why we know only the approximate number of different proteins produced by the human genome.
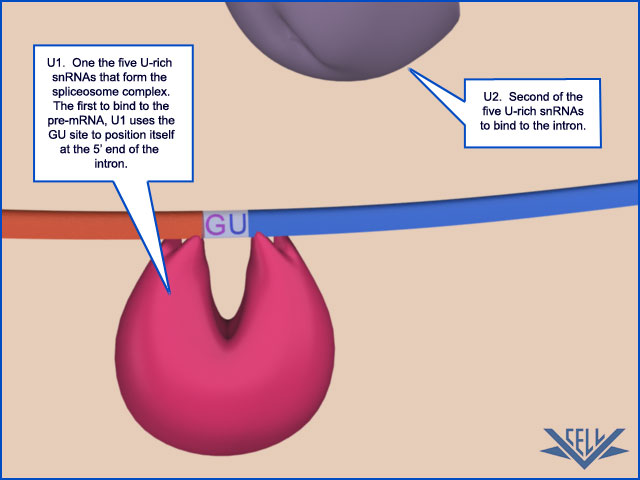
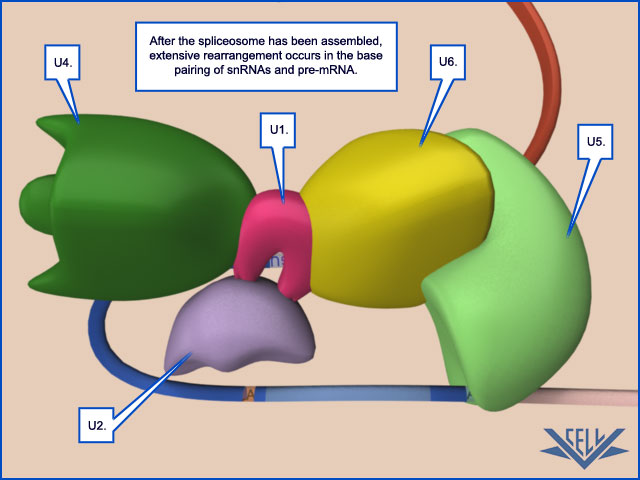
RNA Splicing Is Performed by the Spliceosome
Unlike the other steps of mRNA production we have discussed, key steps in RNA splicing are performed by RNA molecules rather than proteins. Specialized RNA molecules recognize the nucleotide sequences that specify where splicing is to occur and also catalyze the chemistry of splicing. These RNA molecules are relatively short (less than 200 nucleotides each), and there are five of them, U1, U2, U4, U5, and U6. Known as snRNAs (small nuclear RNAs), each is complexed with at least seven protein subunits to form an snRNP (small nuclear ribonucleoprotein).
Question: what good would there be for these Specialized RNA molecules to arise, if there were no machinery to subsequently splice the mRNA's ? even more, the make of the subunits has to be explained as well, since, by their own, there is no function for them
These snRNPs form the core of the spliceosome, the large assembly of RNA and protein molecules that performs pre-mRNA splicing in the cell. During the splicing reaction, recognition of the 5ʹ splice junction, the branch-point site, and the 3ʹ splice junction is performed largely through base-pairing between the snRNAs and the consensus RNA sequences in the pre-mRNA substrate. The spliceosome is a complex and dynamic machine. When studied in vitro, a few components of the spliceosome assemble on pre-mRNA and, as the splicing reaction proceeds, new components enter and those that have already performed their tasks are jettisoned
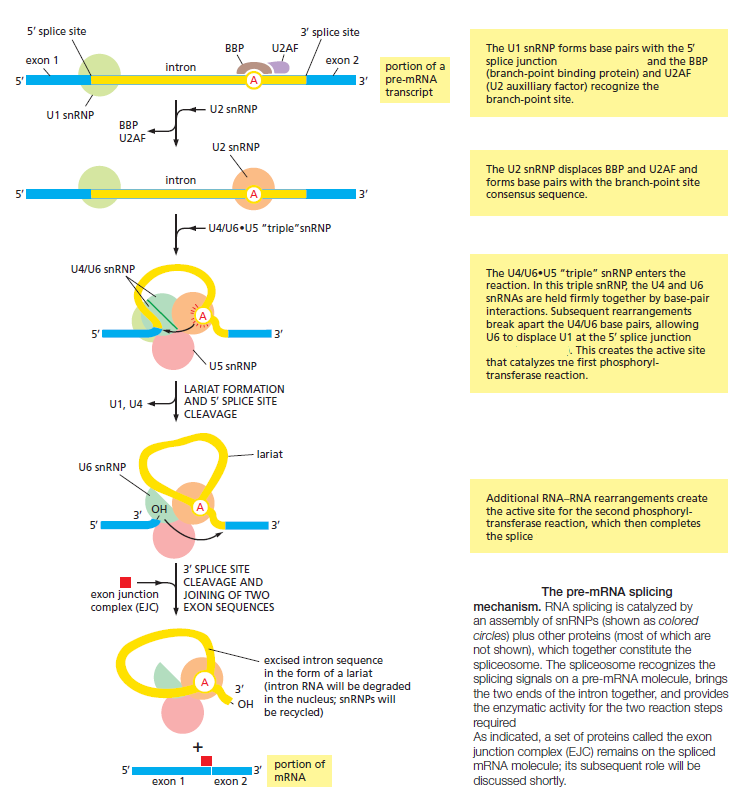
The Spliceosome Uses ATP Hydrolysis to Produce a Complex Series of RNA–RNA Rearrangements
ATP hydrolysis is not required for the chemistry of RNA splicing per se since the two transesterification reactions preserve the high-energy phosphate bonds. However, extensive ATP hydrolysis is required for the assembly and rearrangements of the spliceosome. Some of the additional proteins that make up the spliceosome use the energy of ATP hydrolysis to break existing RNA–RNA interactions to allow the formation of new ones. Each successful splice requires approximately 200 proteins if we include those that form the snRNPs. What is the purpose of these rearrangements? First, they allow the splicing signals on the pre-RNA to be examined by snRNPs several times during the course of
splicing. For example, the U1 snRNP initially recognizes the 5ʹ splice site through conventional base-pairing; as splicing proceeds, these base pairs are broken (using the energy of ATP hydrolysis) and U1 is replaced by U6
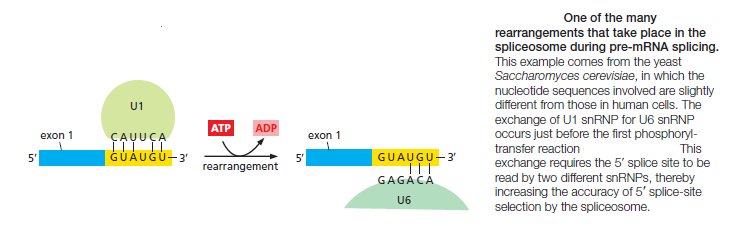
This type of RNA–RNA rearrangement (in which the formation of one RNA–RNA interaction requires the disruption of another) occurs several times during splicing and allows the spliceosomes to check and recheck the splicing signals, thereby increasing the overall accuracy of splicing.
How could this recheck or proofreading have emerged naturally, without a guiding force, without an intelligent designer set up the function with the specific goal to reach a certain, high level of accuracy of the job?
Second, the rearrangements that take place in the spliceosome create the active sites for the two transesterification reactions. These two active sites are created, one after the other, and only after the splicing signals on the pre-mRNA have been checked several times. This orderly progression ensures that splicing accidents occur only rarely. One of the most surprising features of the spliceosome is the nature of the catalytic sites: they are formed by both protein and RNA molecules, although the RNA molecules catalyze the actual chemistry of splicing. In the last section of this chapter, we discuss in general terms the structural and chemical properties of RNA molecules that allow them to act as catalysts. Once the splicing chemistry is completed, the snRNPs remain bound to the lariat. The disassembly of these snRNPs from the lariat (and from each other) requires another series of RNA–RNA rearrangements that require ATP hydrolysis,thereby returning the snRNAs to their original configuration so that they can be used again in a new reaction.
Question: Does the ability to rearrange to exercise new future reactions indicate the planning guide of an intelligent creator? The matter has no specific goals like this, and cannot create these specific goals of repetitive complex functions
At the completion of a splice, the spliceosome directs a set of proteins to bind to the mRNA near the position formerly occupied by the intron. Called the exon junction complex (EJC), these proteins mark the site of a successful splicing event and, as we shall see later in this chapter, influence the subsequent fate of the mRNA.
Other Properties of Pre-mRNA and Its Synthesis Help to Explain the Choice of Proper Splice Sites
Intron sequences vary enormously in size, with some being in excess of 100,000 nucleotides. If splice-site selection were determined solely by the snRNPs acting on a preformed, protein-free RNA molecule, we would expect frequent splicing mistakes—such as exon skipping and the use of “cryptic” splice sites

The fidelity mechanisms built into the spliceosome to suppress errors, however, are supplemented by two additional strategies that further increase the accuracy of splicing. The first is a simple consequence of splicing being coupled to transcription. As transcription proceeds, the phosphorylated tail of RNA polymerase carries several components of the spliceosome and these components are transferred directly from the polymerase to the RNA as the RNA emerges from the polymerase. This strategy helps the cell keep track of introns and exons: for example, the snRNPs that assemble at a 5ʹ splice site are initially presented only with the single 3ʹ splice site that emerges next from the polymerase; the potential sites further downstream have not yet been synthesized. The coordination of transcription with splicing is especially important in preventing inappropriate exon skipping.
A strategy called “exon definition” also helps cells choose the appropriate splice sites. Exon size tends to be much more uniform than intron size, averaging about 150 nucleotide pairs across a wide variety of eukaryotic organisms
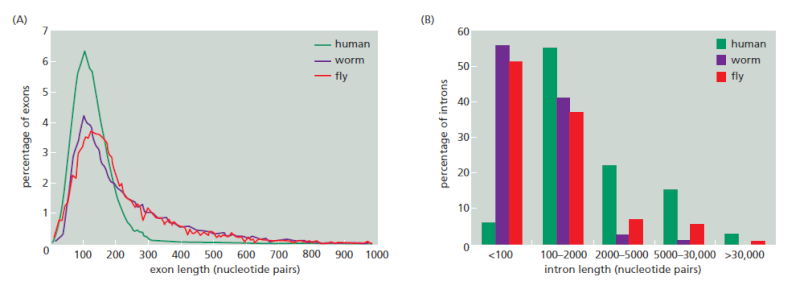
Variation in intron and exon lengths in the human, worm, and fly genomes.
(A) Size distribution of exons.
(B) Size distribution of introns. Note that exon length is much more uniform than intron length.
Through exon definition, the splicing machinery can seek out the relatively homogeneously sized exon sequences. As RNA synthesis proceeds, a group of additional components (most notably SR proteins, so-named because they contain a domain rich in serines and arginines) assemble on exon sequences and help to mark off each 3ʹ and 5ʹ splice site, starting at the 5ʹ end of the RNA
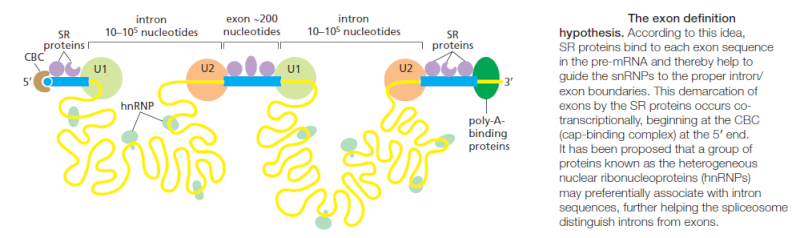
These proteins, in turn, recruit U1 snRNA, which marks the downstream exon boundary, and U2 snRNA, which specifies the upstream one. By specifically marking the exons in this way and thereby taking advantage of the relatively uniform size of exons, the cell increases the accuracy with which it deposits the initial splicing components on the nascent RNA and thereby avoids “near miss” splice sites. How the SR proteins discriminate exon sequences from intron sequences is not understood in detail; however, it is known that some of the SR proteins bind preferentially to specific RNA sequences in exons, termed splicing enhancers. In principle, since any one of several different codons can be used to code for a given amino acid, there is freedom to evolve the exon nucleotide sequence so as to form a binding site for an SR protein, without necessarily affecting the amino acid sequence that the exon specifies. Both the marking of exon and intron boundaries and the assembly of the spliceosome begin on an RNA molecule while it is still being elongated by RNA polymerase at its 3ʹ end. However, the actual chemistry of splicing can take place later. This delay means that intron sequences are not necessarily removed from a premRNA molecule in the order in which they occur along the RNA chain.
Chromatin Structure Affects RNA Splicing
Although it may seem at first counterintuitive, the way a gene is packaged into chromatin can affect how the RNA transcript of that gene is ultimately spliced. Nucleosomes tend to be positioned over exons (which are, on average, close to the length of DNA in a nucleosome), and it has been proposed that these act as “speed bumps,” allowing the proteins responsible for exon definition to assemble on the RNA as it emerges from the polymerase. In addition, changes in chromatin structure are used to alter splicing patterns. There are two ways this can happen. First, because splicing and transcription are coupled, the rate at which RNA polymerase moves along DNA can affect RNA splicing. For example, if polymerase is moving slowly, exon skipping is minimized: assembly of the initial spliceosome may be complete before an alternative choice of splice site even emerges from the RNA polymerase. The nucleosomes in condensed chromatin can cause polymerase to pause; the pattern of pauses in turn affects the extent of RNA exposed at any given time to the splicing machinery. There is a second and more direct way that chromatin structure can affect RNA splicing. Although the details are not yet understood, specific histone modifications attract components of the spliceosome, and, because the chromatin being transcribed is in close association with the nascent RNA, these splicing components can easily be transferred to the emerging RNA. In this way, certain types of histone modifications can affect the final pattern of splicing.
RNA Splicing Shows Remarkable Plasticity
We have seen that the choice of splice sites depends on such features of the premRNA transcript as the strength of the three signals on the RNA (the 5ʹ and 3ʹ splice junctions and the branch point) for the splicing machinery, the co-transcriptional assembly of the spliceosome, chromatin structure, and the “bookkeeping” that underlies exon definition. We do not know exactly how accurate splicing normally is because there are several quality control systems that rapidly destroy mRNAs whose splicing goes awry. However, we do know that, compared with other steps in gene expression, splicing is unusually flexible. Thus, for example, a mutation in a nucleotide sequence critical for splicing of a particular intron does not necessarily prevent splicing of that intron altogether. Instead, the mutation typically creates a new pattern of splicing
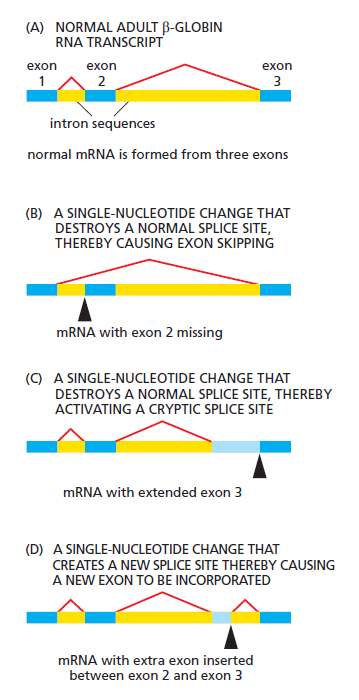
Abnormal processing of the β-globin primary RNA transcript in humans with the disease β thalassemia. In the examples shown, the disease (a severe anemia due to aberrant hemoglobin synthesis) is caused by splice-site mutations found in the genomes of affected patients. The dark blue boxes represent the three normal exon sequences; the red lines connect the 5ʹ and 3ʹ splice sites that are used. In (B), (C), and (D), the light blue boxes depict new nucleotide sequences included in the final mRNA molecule as a result of the mutation denoted by the black arrowhead. Note that when a mutation leaves a normal splice site without a partner, an exon is skipped (B) or one or more abnormal cryptic splice sites nearby is used as the partner site (C).
Most commonly, an exon is simply skipped (Figure B). In other cases, the mutation causes a cryptic splice junction to be efficiently used (Figure C). Apparently, the splicing machinery does pick out the best possible pattern of splice junctions, and if the optimal one is damaged by mutation, it will seek out the next best pattern, and so on. This inherent plasticity in the process of RNA splicing suggests that changes in splicing patterns caused by random mutations are important for genes and organisms. It also means that mutations that affect splicing can be severely detrimental to the organism: in addition to the β thalassemia, example presented in the Figure above, aberrant splicing plays important roles in the development of cystic fibrosis, frontotemporal dementia, Parkinson’s disease, retinitis pigmentosa, spinal muscular atrophy, myotonic dystrophy, premature aging, and cancer. It has been estimated that of the many point mutations that cause inherited human diseases, 10% produce aberrant splicing of the gene containing the mutation.
The plasticity of RNA splicing also means that the cell can easily regulate the pattern of RNA splicing. Alternative splicing can give rise to different proteins from the same gene and that this is a common strategy to enhance the coding potential of genomes. Some examples of alternative splicing are constitutive; that is, the alternatively spliced mRNAs are produced continuously by cells of an organism. However, in many cases, the cell regulates the splicing patterns so that different forms of the protein are produced at different
times and in different tissues .
Spliceosome-Catalyzed RNA Splicing Probably emerged from Self-splicing Mechanisms
When the spliceosome was first discovered, it puzzled molecular biologists. Why do RNA molecules instead of proteins perform important roles in splice-site recognition and in the chemistry of splicing? Why is a lariat intermediate used rather than the apparently simpler alternative of bringing the 5ʹ and 3ʹ splice sites together in a single step, followed by their direct cleavage and rejoining? It is likely that early cells used RNA molecules rather than proteins as their major catalysts and that they stored their genetic information in RNA rather than in DNA sequences. RNA-catalyzed splicing reactions presumably had critical roles in these early cells. As evidence, some self-splicing RNA introns (that is, intron sequences in RNA whose splicing out can occur in the absence of proteins or any other RNA molecules) remain today—for example, in the nuclear rRNA genes of the ciliate Tetrahymena, in a few bacteriophage T4 genes, and in some mitochondrial and chloroplast genes. In these cases, the RNA molecule folds into a specific three-dimensional structure that brings the intron/exon junctions together and catalyzes the two transesterification reactions. A self-splicing intron sequence can be identified in a test tube by incubating a pure RNA molecule that contains the intron sequence and observing the splicing reaction. Because the basic chemistry of some self-splicing reactions is so similar to pre-mRNA splicing, it has been proposed that the much more involved process of pre-mRNA splicing evolved from a simpler, ancestral form of RNA self-splicing
RNA-Processing Enzymes Generate the 3ʹ End of Eukaryotic mRNAs
We have seen that the 5ʹ end of the pre-mRNA produced by RNA polymerase II is capped almost as soon as it emerges from the RNA polymerase. Then, as the polymerase continues its movement along a gene, the spliceosome assembles on the RNA and delineates the intron and exon boundaries. The long C-terminal tail of the RNA polymerase coordinates these processes by transferring capping and splicing components directly to the RNA as it emerges from the enzyme. In this section, we shall see that, as RNA polymerase II reaches the end of a gene, a similar mechanism ensures that the 3ʹ end of the pre-mRNA is appropriately processed. The position of the 3ʹ end of each mRNA molecule is specified by signals encoded in the genome
1) http://reasonandscience.heavenforum.org/t2022-3-end-cleavage-and-polyadenylation
More:
Protein recipe requires precise timing
Molecular architecture of the human U4/U6.U5 tri-snRNP
https://reasonandscience.catsboard.com/t2180-the-spliceosome-the-splicing-code-and-pre-mrna-processing-in-eukaryotic-cells
Along the way to make proteins in eukaryotic cells, there is a whole chain of subsequent events that must all be fully operational, and the machinery in place, in order to get the functional product, that is proteins. At the beginning of the process, DNA is transcribed in the RNA polymerase molecular machine, to yield messenger RNA ( mRNA ), which afterward must go through post-transcriptional modifications. That is CAPPING, ELONGATION, SPLICING, CLEAVAGE, POLYADENYLATION, AND TERMINATION before it can be EXPORTED FROM THE NUCLEUS TO THE CYTOSOL, and PROTEIN SYNTHESIS INITIATED, (TRANSLATION), and COMPLETION OF PROTEIN SYNTHESIS AND PROTEIN FOLDING.
Bacterial mRNAs are synthesized by the RNA polymerase starting and stopping at specific spots on the genome. The situation in eukaryotes is substantially different. In particular, transcription is only the first of several steps needed to produce a mature mRNA molecule. The mature transcript for many genes is encoded in a discontinuous manner in a series of discrete exons, which are separated from each other along the DNA strand by non-coding introns. mRNAs, rRNAs, and tRNAs can all contain introns that must be removed from precursor RNAs to produce functional molecules.The formidable task of identifying and splicing together exons among all the intronic RNA is performed by a large ribonucleoprotein machine, the spliceosome, which is composed of several individual small nuclear ribonucleoproteins, five snRNPs, pronounced ‘snurps’, (U1, U2, U4, U5, and U6) each containing an RNA molecule called an snRNA that is usually 100–300 nucleotides long, plus additional protein factors that recognize specific sequences in the mRNA or promote conformational rearrangements in the spliceosome required for the splicing reaction to progress, and many more additional proteins that come and go during the splicing reaction. It has been described as one of "the most complex macromolecular machines known," "composed of as many as 300 distinct proteins and five RNAs".
The snRNAs perform many of the spliceosome’s mRNA recognition events. Splice site consensus sequences are recognized by non-snRNP factors; the branch-point sequence is recognized by the branch-point-binding protein (BBP), and the polypyrimidine tract and 3′ splice site are bound by two specific protein components of a splicing complex referred to as U2AF (U2 auxiliary factor), U2AF65 and U2AF35, respectively.
This is one more great example of an amazingly complex molecular machine, that will operate and exercise its precise orchestrated function properly ONLY with ALL components fully developed and formed and able to interact in a highly complex, ordered precise manner. Both, the software, and the hardware must be in place fully developed, or the machine will not work. No intermediate stage will do the job. And neither would snRNPs (U1, U2, U4, U5, and U6) have any function if not fully developed. And even if they were there, without the branch-point-binding protein (BBP) in place, nothing done, either, since the correct splice site could not be recognized. Had the introns and exons not have to emerge simultaneously with the Spliceosome? No wonder, does the paper: " Origin and evolution of spliceosomal introns " admit: Evolution of the exon-intron structure of eukaryotic genes has been a matter of long-standing, intensive debate. 1 and it concludes that: The elucidation of the general scenario of the evolution of eukaryote gene architecture by no account implies that the main problems in the study of intron evolution and function have been solved. Quite the contrary, fundamental questions remain wide open. If the first evolutionary step would have been the arise of self-splicing Group II introns, then the question would follow: Why would evolution not have stopped there, since that method works just fine?
There is no credible road map, how introns and exons, and the splice function could have emerged gradually. What good would the spliceosome be good for, if the essential sequence elements to recognize where to slice would not be in place? What would happen, if the pre mRNA with exons and introns were in place, but no spliceosome ready in place to do the post-transcriptional modification, and neither the splicing code, which directs the way where to splice? In the article: ‘JUNK’ DNA HIDES ASSEMBLY INSTRUCTIONS, the author, Wang, observes that splicing "is a tightly regulated process, and a great number of diseases are caused by the 'misregulation' of splicing in which the gene was not cut and pasted correctly." Missplicing in the cell can have dire consequences as the desired product is not produced, and often the wrong products can be toxic for the cell. For this reason, it has been proposed that ATPases are important for ‘proofreading’ mechanisms that promote fidelity in splice site selection.
In his textbook Essentials of Molecular Biology, George Malacinski points out why proper polypeptide production is critical:
"A cell cannot, of course, afford to miss any of the splice junctions by even a single nucleotide, because this could result in an interruption of the correct reading frame, leading to a truncated protein."
The required precision is quite amazing, and even more astounding is the fact that these incredibly complex molecular machines are able and capable to do the job in the precise manner as needed.
Following the binding of these initial components, the remainder of the splicing apparatus assembles around them, in some cases displacing some of the previously bound components.
Question: How could the information to assemble the splicing apparatus correctly have emerged gradually? In order to do so, had the assembly parts not have to be there, at the assembly site, fully developed, and ready for recruitment? Had the availability of these parts not have to be synchronized so that at some point, either individually or in combination, they were all available at the same time? Had the assembly not have to be coordinated in the right way right from the start? Had the parts not have to be mutually compatible, that is, ‘well-matched’ and capable of properly ‘interacting’? even if subsystems or parts are put together in the right order, they also need to interface correctly.
Is it feasible that this complex machine was the result of progressive evolutionary development, in which simple molecules are the start of the biosynthesis chain and are then progressively developed in sequential steps if the end goal is not known by the process and mechanism promoting the development? How could each intermediate in the pathway be an endpoint in the pathway, if that endpoint had no function? Did not each intermediate have to be usable in the past as an end product? And how could the be usable, if the amino acid sequence chain had only a fraction of the fully developed sequence? How could successive steps be added to improve the efficiency of a product where there was no use for it at this stage? Despite the fact that proponents of naturalism embrace this kind of scenario, it seems obvious that is extremely unlikely to be possible that way.
Martin and Koonin admit in their paper “Hypothesis: Introns and the origin of nucleus-cytosol compartmentalization,”: The transition to spliceosome-dependent splicing will also impose an unforgiving demand for inventions in addition to the spliceosome. And furthermore: More recent are the insights that there is virtually no evolutionary grade detectable in the origin of the spliceosome, which apparently was present in its (almost) fully-fledged state in the common ancestor of eukaryotic lineages studied so far. That's a surprising admittance.
This means that the spliceosome appeared fully formed almost abruptly and that the intron invasion took place over a short time and has not changed for supposedly hundreds of millions of years.
In another interesting paper : Breaking the second genetic code, the authors write 2 : The genetic instructions of complex organisms exhibit a counter-intuitive feature not shared by simpler genomes: nucleotide sequences coding for a protein (exons) are interrupted by other nucleotide regions that seem to hold no information (introns). This bizarre organization of genetic messages forces cells to remove introns from the precursor mRNA (pre-mRNA) and then splice together the exons to generate translatable instructions. An advantage of this mechanism is that it allows different cells to choose alternative means of pre-mRNA splicing and thus generates diverse messages from a single gene. The variant mRNAs can then encode different proteins
with distinct functions. One difficulty with understanding alternative pre-mRNA splicing is that the selection of particular exons in mature mRNAs is determined not only by intron sequences adjacent to the exon boundaries but also by a multitude of other sequence elements present in both exons and introns. These auxiliary sequences are recognized by regulatory factors that assist or prevent the function of the spliceosome — the molecular machinery in charge of intron removal.
Moreover, the coupling between RNA processing and gene transcription influences alternative splicing, and recent data implicate the packing of DNA with histone proteins and histone covalent modifications — the epigenetic code — in the regulation of splicing. The interplay between the histone and the splicing codes will, therefore, need to be accurately formulated in future approaches.
Question: How could natural mechanisms have provided the tuning, synchronization, and coordination between the histone and the splicing codes? First, these two codes and the carrier proteins and molecules ( the hardware and software ) would have to emerge by themselves, and in a second step orchestrate their coordination. Why is it reasonable to believe, that unguided, random chemical reactions would be capable of emerging with the immensely complex organismal functions?
Fazale Rana puts it nicely: Astounding is the fact that other codes, such as the histone binding code, transcription factor binding code, the splicing code, and the RNA secondary structure code, overlap the genetic code. Each of these codes plays a special role in gene expression, but they also must work together in a coherent integrated fashion.
1) http://www.biologydirect.com/content/7/1/11
2) http://nar.oxfordjournals.org/content/early/2013/11/07/nar.gkt1053.full.pdf
The spliceosome, the splicing code, and pre - mRNA processing in eukaryotic cells
Transcription Elongation in Eukaryotes Is Tightly Coupled to RNA Processing
Bacterial mRNAs are synthesized by the RNA polymerase starting and stopping at specific spots on the genome. The situation in eukaryotes is substantially different. In particular, transcription is only the first of several steps needed to produce a mature mRNA molecule. Other critical steps are the covalent modification of the ends of the RNA and the removal of intron sequences that are discarded from the middle of the RNA transcript by the process of RNA splicing

Comparison of the steps leading from gene to protein in eukaryotes and bacteria. The final level of a protein in the cell depends on the efficiency of each step and on the rates of degradation of the RNA and protein molecules.
(A) In eukaryotic cells, the mRNA molecule resulting from transcription contains both coding (exon) and noncoding (intron) sequences. Before it can be translated into protein, the two ends of the RNA are modified, the introns are removed by an enzymatically catalyzed RNA splicing reaction, and the resulting mRNA is transported from the nucleus to the cytoplasm. For convenience, the steps in this figure are depicted as occurring one at a time; in reality, many occur concurrently. For example, the RNA cap is added and splicing begins before transcription has been completed. Because of the coupling between transcription and RNA processing,
intact primary transcripts—the full-length RNAs that would, in theory, be produced if no processing had occurred—are found only rarely.
(B) In prokaryotes, the production of mRNA is much simpler. The 5ʹ end of an mRNA molecule is produced by the initiation of transcription, and the 3ʹ end is produced by the termination of transcription. Since prokaryotic cells lack a nucleus, transcription and translation take place in a common compartment, and the translation of a bacterial mRNA often begins before its synthesis has been completed.
Both ends of eukaryotic mRNAs are modified: by capping on the 5ʹ end and by polyadenylation of the 3ʹ end 1 These special ends allow the cell to assess whether both ends of a mRNA molecule are present (and if the message is therefore intact) before it exports the RNA from the nucleus and translates it into protein.
Question: How could and would unguided, natural processes, where no intelligence is involved, emerge with that check out if both ends of mRNA are present? Quality control is usually an exercise attributed to intelligence.
RNA splicing joins together the different portions of a protein-coding sequence, and it provides eukaryotes with the ability to synthesize several different proteins from the same gene. A simple strategy
Why not designed? figuring out what strategy to apply to get the best results is a mental process
to couple all of the above RNA processing steps to transcription elongation. As discussed previously, a key step in transcription initiation by RNA polymerase II is the phosphorylation of the RNA polymerase II tail, also called the CTD (C-terminal domain). This phosphorylation, which proceeds gradually as the RNA polymerase initiates transcription and moves along the DNA, not only helps dissociate the RNA polymerase II from other proteins present at the start point of transcription, but also allows a new set of proteins to associate with the RNA polymerase tail that function in transcription elongation and RNA processing. As discussed next, some of these processing proteins are thought to “hop” from the polymerase tail onto the nascent RNA molecule to begin processing it as it emerges from the RNA polymerase. Thus, we can view RNA polymerase II in its elongation mode as an RNA factory that not only moves along the DNA synthesizing an RNA molecule but also processes the RNA that it produces

Fully extended, the CTD is nearly 10 times longer than the remainder of RNA polymerase. As a flexible protein domain, it serves as a scaffold or tether, holding a variety of proteins close by so that they can rapidly act when needed. This strategy, which greatly speeds up the overall rate of a series of consecutive reactions, is one that is commonly utilized in the cell.
RNA Capping Is the First Modification of Eukaryotic Pre-mRNAs
As soon as RNA polymerase II has produced about 25 nucleotides of RNA, the 5ʹ end of the new RNA molecule is modified by addition of a cap that consists of a modified guanine nucleotide. Three enzymes, acting in succession, perform the capping reaction: one (a phosphatase) removes a phosphate from the 5ʹ end of the nascent RNA, another (a guanyl transferase) adds a GMP in a reverse linkage (5ʹ to 5ʹ instead of 5ʹ to 3ʹ), and a third (a methyl transferase) adds a methyl group to the guanosine

The reactions that cap the 5ʹ end of each RNA molecule synthesized by RNA polymerase II. The final cap contains a novel 5ʹ-to-5ʹ linkage between the positively charged 7-methyl G residue and the 5ʹ end of the RNA transcript . The letter N represents any one of the four ribonucleotides, although the nucleotide that starts an RNA chain is usually a purine (an A or a G).
Because all three enzymes bind to the RNA polymerase tail phosphorylated at the Ser5 position—the modification added by TFIIH during transcription initiation— they are poised to modify the 5ʹ end of the nascent transcript as soon as it emerges from the polymerase. The 5ʹ-methyl cap signifies the 5ʹ end of eukaryotic mRNAs, and this landmark helps the cell to distinguish mRNAs from the other types of RNA molecules present in the cell. For example, RNA polymerases I and III produce uncapped RNAs during transcription, in part because these polymerases lack a CTD. In the nucleus, the cap binds a protein complex called CBC (cap-binding complex), which, as we discuss in subsequent sections, helps a future mRNA be further processed and exported. The 5ʹ-methyl cap also has an important role in the translation of mRNAs in the cytosol, as we discuss later in the chapter.
RNA Splicing Removes Intron Sequences from Newly Transcribed Pre-mRNAs
The protein-coding sequences of eukaryotic genes are typically interrupted by noncoding intervening sequences (introns). Discovered in 1977, this feature of eukaryotic genes came as a surprise to scientists, who had been, until that time, familiar only with bacterial genes, which typically consist of a continuous stretch of coding DNA that is directly transcribed into mRNA. In marked contrast, eukaryotic genes were found to be broken up into small pieces of coding sequence (expressed sequences or exons) interspersed with much longer intervening sequences or introns; thus, the coding portion of a eukaryotic gene is often only a small fraction of the length of the gene

Structure of two human genes showing the arrangement of exons and introns.
(A) The relatively small β-globin gene, which encodes a subunit of the oxygen-carrying protein hemoglobin, contains 3 exons .
(B) The much larger Factor VIII gene contains 26 exons; it codes for a protein (Factor VIII) that functions in the bloodclotting pathway. The most prevalent form of hemophilia results from mutations in this gene.
Both intron and exon sequences are transcribed into RNA. The intron sequences are removed from the newly synthesized RNA through the process of RNA splicing. The vast majority of RNA splicing that takes place in cells functions in the production of mRNA, and our discussion of splicing focuses on this so-called precursor-mRNA (or pre-mRNA) splicing. Only after 5ʹ- and 3ʹ-end processing and splicing have taken place is such RNA termed mRNA. Each splicing event removes one intron, proceeding through two sequential
phosphoryl-transfer reactions known as transesterifications; these join two exons together while removing the intron between them as a “lariat”

This is a highly ordered, sequential, and precise mechanism. How did this function emerge naturally? How was the specific adenine nucleotide put into the right place to be spliced? trial and error? In order to propose a natural origin, these questions need to be explained in a compelling manner.
The machinery that catalyzes pre-mRNA splicing is complex, consisting of five additional RNA molecules and several hundred proteins, and it hydrolyzes many ATP molecules per splicing event. This complexity ensures that splicing is accurate, while at the same time being flexible enough to deal with the enormous variety of introns found in a typical eukaryotic cell. It may seem wasteful to remove large numbers of introns by RNA splicing. In attempting to explain why it occurs, scientists have pointed out that the exon-intron arrangement would seem to facilitate the emergence of new and useful proteins over evolutionary timescales.
First of all, proponents of evolution should be able to explain why such a piece of complex machinery would emerge AT ALL, and why there should be a need of emergence of such complexity if bacterial cells work just fine without it. What I see here, is typical simplistic assertions without giving a considerable thought about the viability of random mutations to produce such enormous complexity
Thus, the presence of numerous introns in DNA allows genetic recombination to readily combine the exons of different genes, enabling genes for new proteins to
Why should nature give preference to such a completely new method of producing a variety of proteins, rather than just let mutations and natural selection doing the job? It seems to me, in order to keep the pre-established fact of evolution, their proponents simply shut up with their critical thinking, and accept the fact of evolution, no matter how unlikely it seems

Rather than being the wasteful process it may have seemed at first sight, RNA splicing enables eukaryotes to increase the coding potential of their genomes.
In order to splice out the introns from exons, the cell required epigenetic information beside the complex machinery in place to do this complex task. Where did this information come from? trial and error?
Nucleotide Sequences Signal Where Splicing Occurs
The mechanism of pre-mRNA splicing requires that the splicing machinery recognize three portions of the precursor RNA molecule: the 5ʹ splice site, the 3ʹ splice site, and the branch point in the intron sequence that forms the base of the excised lariat. Not surprisingly, each site has a consensus nucleotide sequence that is similar from intron to intron and provides the cell with cues for where splicing is to take place.

The consensus nucleotide sequences in an RNA molecule that signal the beginning and the end of most introns in humans. The three blocks of nucleotide sequences shown are required to remove an intron sequence. Here A, G, U, and C are the standard RNA nucleotides; R stands for purines (A or G); and Y stands for pyrimidines (C or U). The A highlighted in red forms the branch point of the lariat produced by splicing. Only the GU at the start of the intron and the AG at its end are invariant nucleotides in the splicing consensus sequences. Several different nucleotides can occupy the remaining positions, although the indicated nucleotides are preferred. The distances along the RNA between the three splicing consensus sequences are highly variable; however, the distance between the branch point and 3ʹ splice junction is typically much shorter than that between the 5ʹ splice junction and the branch point.
However, these consensus sequences are relatively short and can accommodate extensive sequence variability; the cell incorporates additional types of information to ultimately choose exactly where, on each RNA molecule, splicing is to take place.
Its evident that foresight is required a) to make the splicing machinery, and b) to program the information right from the start, where to slice; the whole mechanism has to arise all at once, fully functional. A stepwise arise is extremely unlikely, not to say, impossible. What good would the spliceosome be good for, if there were no information at the same time in order to make the machinery do its job? The hardware and the software are interdependent.
The high variability of the splicing consensus sequences presents a special challenge for scientists attempting to decipher genome sequences. Introns range in size from about 10 nucleotides to over 100,000 nucleotides, and choosing the precise borders of each intron is a difficult task even with the aid of powerful computers.
But cells " know " where to splice for a long time..... how did they " learn " that feat?
The possibility of alternative splicing compounds the problem of predicting protein sequences solely from a genome sequence. This difficulty is one of the main barriers to identifying all of the genes in a complete genome sequence, and it is one of the primary reasons why we know only the approximate number of different proteins produced by the human genome.
RNA Splicing Is Performed by the Spliceosome
Unlike the other steps of mRNA production we have discussed, key steps in RNA splicing are performed by RNA molecules rather than proteins. Specialized RNA molecules recognize the nucleotide sequences that specify where splicing is to occur and also catalyze the chemistry of splicing. These RNA molecules are relatively short (less than 200 nucleotides each), and there are five of them, U1, U2, U4, U5, and U6. Known as snRNAs (small nuclear RNAs), each is complexed with at least seven protein subunits to form an snRNP (small nuclear ribonucleoprotein).
Question: what good would there be for these Specialized RNA molecules to arise, if there were no machinery to subsequently splice the mRNA's ? even more, the make of the subunits has to be explained as well, since, by their own, there is no function for them
These snRNPs form the core of the spliceosome, the large assembly of RNA and protein molecules that performs pre-mRNA splicing in the cell. During the splicing reaction, recognition of the 5ʹ splice junction, the branch-point site, and the 3ʹ splice junction is performed largely through base-pairing between the snRNAs and the consensus RNA sequences in the pre-mRNA substrate. The spliceosome is a complex and dynamic machine. When studied in vitro, a few components of the spliceosome assemble on pre-mRNA and, as the splicing reaction proceeds, new components enter and those that have already performed their tasks are jettisoned

The Spliceosome Uses ATP Hydrolysis to Produce a Complex Series of RNA–RNA Rearrangements
ATP hydrolysis is not required for the chemistry of RNA splicing per se since the two transesterification reactions preserve the high-energy phosphate bonds. However, extensive ATP hydrolysis is required for the assembly and rearrangements of the spliceosome. Some of the additional proteins that make up the spliceosome use the energy of ATP hydrolysis to break existing RNA–RNA interactions to allow the formation of new ones. Each successful splice requires approximately 200 proteins if we include those that form the snRNPs. What is the purpose of these rearrangements? First, they allow the splicing signals on the pre-RNA to be examined by snRNPs several times during the course of
splicing. For example, the U1 snRNP initially recognizes the 5ʹ splice site through conventional base-pairing; as splicing proceeds, these base pairs are broken (using the energy of ATP hydrolysis) and U1 is replaced by U6

This type of RNA–RNA rearrangement (in which the formation of one RNA–RNA interaction requires the disruption of another) occurs several times during splicing and allows the spliceosomes to check and recheck the splicing signals, thereby increasing the overall accuracy of splicing.
How could this recheck or proofreading have emerged naturally, without a guiding force, without an intelligent designer set up the function with the specific goal to reach a certain, high level of accuracy of the job?
Second, the rearrangements that take place in the spliceosome create the active sites for the two transesterification reactions. These two active sites are created, one after the other, and only after the splicing signals on the pre-mRNA have been checked several times. This orderly progression ensures that splicing accidents occur only rarely. One of the most surprising features of the spliceosome is the nature of the catalytic sites: they are formed by both protein and RNA molecules, although the RNA molecules catalyze the actual chemistry of splicing. In the last section of this chapter, we discuss in general terms the structural and chemical properties of RNA molecules that allow them to act as catalysts. Once the splicing chemistry is completed, the snRNPs remain bound to the lariat. The disassembly of these snRNPs from the lariat (and from each other) requires another series of RNA–RNA rearrangements that require ATP hydrolysis,thereby returning the snRNAs to their original configuration so that they can be used again in a new reaction.
Question: Does the ability to rearrange to exercise new future reactions indicate the planning guide of an intelligent creator? The matter has no specific goals like this, and cannot create these specific goals of repetitive complex functions
At the completion of a splice, the spliceosome directs a set of proteins to bind to the mRNA near the position formerly occupied by the intron. Called the exon junction complex (EJC), these proteins mark the site of a successful splicing event and, as we shall see later in this chapter, influence the subsequent fate of the mRNA.
Other Properties of Pre-mRNA and Its Synthesis Help to Explain the Choice of Proper Splice Sites
Intron sequences vary enormously in size, with some being in excess of 100,000 nucleotides. If splice-site selection were determined solely by the snRNPs acting on a preformed, protein-free RNA molecule, we would expect frequent splicing mistakes—such as exon skipping and the use of “cryptic” splice sites

The fidelity mechanisms built into the spliceosome to suppress errors, however, are supplemented by two additional strategies that further increase the accuracy of splicing. The first is a simple consequence of splicing being coupled to transcription. As transcription proceeds, the phosphorylated tail of RNA polymerase carries several components of the spliceosome and these components are transferred directly from the polymerase to the RNA as the RNA emerges from the polymerase. This strategy helps the cell keep track of introns and exons: for example, the snRNPs that assemble at a 5ʹ splice site are initially presented only with the single 3ʹ splice site that emerges next from the polymerase; the potential sites further downstream have not yet been synthesized. The coordination of transcription with splicing is especially important in preventing inappropriate exon skipping.
A strategy called “exon definition” also helps cells choose the appropriate splice sites. Exon size tends to be much more uniform than intron size, averaging about 150 nucleotide pairs across a wide variety of eukaryotic organisms

Variation in intron and exon lengths in the human, worm, and fly genomes.
(A) Size distribution of exons.
(B) Size distribution of introns. Note that exon length is much more uniform than intron length.
Through exon definition, the splicing machinery can seek out the relatively homogeneously sized exon sequences. As RNA synthesis proceeds, a group of additional components (most notably SR proteins, so-named because they contain a domain rich in serines and arginines) assemble on exon sequences and help to mark off each 3ʹ and 5ʹ splice site, starting at the 5ʹ end of the RNA

These proteins, in turn, recruit U1 snRNA, which marks the downstream exon boundary, and U2 snRNA, which specifies the upstream one. By specifically marking the exons in this way and thereby taking advantage of the relatively uniform size of exons, the cell increases the accuracy with which it deposits the initial splicing components on the nascent RNA and thereby avoids “near miss” splice sites. How the SR proteins discriminate exon sequences from intron sequences is not understood in detail; however, it is known that some of the SR proteins bind preferentially to specific RNA sequences in exons, termed splicing enhancers. In principle, since any one of several different codons can be used to code for a given amino acid, there is freedom to evolve the exon nucleotide sequence so as to form a binding site for an SR protein, without necessarily affecting the amino acid sequence that the exon specifies. Both the marking of exon and intron boundaries and the assembly of the spliceosome begin on an RNA molecule while it is still being elongated by RNA polymerase at its 3ʹ end. However, the actual chemistry of splicing can take place later. This delay means that intron sequences are not necessarily removed from a premRNA molecule in the order in which they occur along the RNA chain.
Chromatin Structure Affects RNA Splicing
Although it may seem at first counterintuitive, the way a gene is packaged into chromatin can affect how the RNA transcript of that gene is ultimately spliced. Nucleosomes tend to be positioned over exons (which are, on average, close to the length of DNA in a nucleosome), and it has been proposed that these act as “speed bumps,” allowing the proteins responsible for exon definition to assemble on the RNA as it emerges from the polymerase. In addition, changes in chromatin structure are used to alter splicing patterns. There are two ways this can happen. First, because splicing and transcription are coupled, the rate at which RNA polymerase moves along DNA can affect RNA splicing. For example, if polymerase is moving slowly, exon skipping is minimized: assembly of the initial spliceosome may be complete before an alternative choice of splice site even emerges from the RNA polymerase. The nucleosomes in condensed chromatin can cause polymerase to pause; the pattern of pauses in turn affects the extent of RNA exposed at any given time to the splicing machinery. There is a second and more direct way that chromatin structure can affect RNA splicing. Although the details are not yet understood, specific histone modifications attract components of the spliceosome, and, because the chromatin being transcribed is in close association with the nascent RNA, these splicing components can easily be transferred to the emerging RNA. In this way, certain types of histone modifications can affect the final pattern of splicing.
RNA Splicing Shows Remarkable Plasticity
We have seen that the choice of splice sites depends on such features of the premRNA transcript as the strength of the three signals on the RNA (the 5ʹ and 3ʹ splice junctions and the branch point) for the splicing machinery, the co-transcriptional assembly of the spliceosome, chromatin structure, and the “bookkeeping” that underlies exon definition. We do not know exactly how accurate splicing normally is because there are several quality control systems that rapidly destroy mRNAs whose splicing goes awry. However, we do know that, compared with other steps in gene expression, splicing is unusually flexible. Thus, for example, a mutation in a nucleotide sequence critical for splicing of a particular intron does not necessarily prevent splicing of that intron altogether. Instead, the mutation typically creates a new pattern of splicing

Abnormal processing of the β-globin primary RNA transcript in humans with the disease β thalassemia. In the examples shown, the disease (a severe anemia due to aberrant hemoglobin synthesis) is caused by splice-site mutations found in the genomes of affected patients. The dark blue boxes represent the three normal exon sequences; the red lines connect the 5ʹ and 3ʹ splice sites that are used. In (B), (C), and (D), the light blue boxes depict new nucleotide sequences included in the final mRNA molecule as a result of the mutation denoted by the black arrowhead. Note that when a mutation leaves a normal splice site without a partner, an exon is skipped (B) or one or more abnormal cryptic splice sites nearby is used as the partner site (C).
Most commonly, an exon is simply skipped (Figure B). In other cases, the mutation causes a cryptic splice junction to be efficiently used (Figure C). Apparently, the splicing machinery does pick out the best possible pattern of splice junctions, and if the optimal one is damaged by mutation, it will seek out the next best pattern, and so on. This inherent plasticity in the process of RNA splicing suggests that changes in splicing patterns caused by random mutations are important for genes and organisms. It also means that mutations that affect splicing can be severely detrimental to the organism: in addition to the β thalassemia, example presented in the Figure above, aberrant splicing plays important roles in the development of cystic fibrosis, frontotemporal dementia, Parkinson’s disease, retinitis pigmentosa, spinal muscular atrophy, myotonic dystrophy, premature aging, and cancer. It has been estimated that of the many point mutations that cause inherited human diseases, 10% produce aberrant splicing of the gene containing the mutation.
The plasticity of RNA splicing also means that the cell can easily regulate the pattern of RNA splicing. Alternative splicing can give rise to different proteins from the same gene and that this is a common strategy to enhance the coding potential of genomes. Some examples of alternative splicing are constitutive; that is, the alternatively spliced mRNAs are produced continuously by cells of an organism. However, in many cases, the cell regulates the splicing patterns so that different forms of the protein are produced at different
times and in different tissues .
Spliceosome-Catalyzed RNA Splicing Probably emerged from Self-splicing Mechanisms
When the spliceosome was first discovered, it puzzled molecular biologists. Why do RNA molecules instead of proteins perform important roles in splice-site recognition and in the chemistry of splicing? Why is a lariat intermediate used rather than the apparently simpler alternative of bringing the 5ʹ and 3ʹ splice sites together in a single step, followed by their direct cleavage and rejoining? It is likely that early cells used RNA molecules rather than proteins as their major catalysts and that they stored their genetic information in RNA rather than in DNA sequences. RNA-catalyzed splicing reactions presumably had critical roles in these early cells. As evidence, some self-splicing RNA introns (that is, intron sequences in RNA whose splicing out can occur in the absence of proteins or any other RNA molecules) remain today—for example, in the nuclear rRNA genes of the ciliate Tetrahymena, in a few bacteriophage T4 genes, and in some mitochondrial and chloroplast genes. In these cases, the RNA molecule folds into a specific three-dimensional structure that brings the intron/exon junctions together and catalyzes the two transesterification reactions. A self-splicing intron sequence can be identified in a test tube by incubating a pure RNA molecule that contains the intron sequence and observing the splicing reaction. Because the basic chemistry of some self-splicing reactions is so similar to pre-mRNA splicing, it has been proposed that the much more involved process of pre-mRNA splicing evolved from a simpler, ancestral form of RNA self-splicing
RNA-Processing Enzymes Generate the 3ʹ End of Eukaryotic mRNAs
We have seen that the 5ʹ end of the pre-mRNA produced by RNA polymerase II is capped almost as soon as it emerges from the RNA polymerase. Then, as the polymerase continues its movement along a gene, the spliceosome assembles on the RNA and delineates the intron and exon boundaries. The long C-terminal tail of the RNA polymerase coordinates these processes by transferring capping and splicing components directly to the RNA as it emerges from the enzyme. In this section, we shall see that, as RNA polymerase II reaches the end of a gene, a similar mechanism ensures that the 3ʹ end of the pre-mRNA is appropriately processed. The position of the 3ʹ end of each mRNA molecule is specified by signals encoded in the genome
1) http://reasonandscience.heavenforum.org/t2022-3-end-cleavage-and-polyadenylation
More:
Protein recipe requires precise timing
Molecular architecture of the human U4/U6.U5 tri-snRNP
Last edited by Admin on Sat Jul 11, 2020 3:05 am; edited 12 times in total