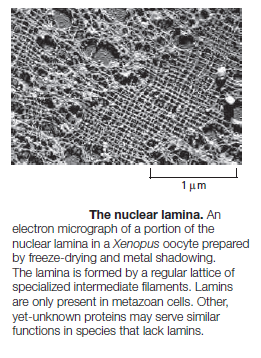
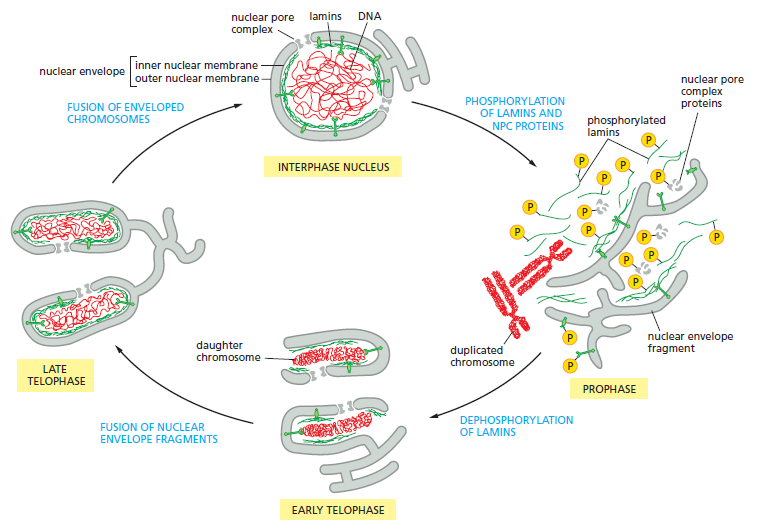
The Nuclear Lamins
The type V intermediate filament proteins are the nuclear lamins. These are considered to be nucleoskeletal IF proteins that are determinants of nuclear size, shape, mechanical integrity, and positioning of nuclear pores. They also play important roles in DNA replication, transcription and the disassembly of the nucleus in mitosis and the reassembly of the nucleus in daughter cells.
Lamins are are concentrated in the nuclear lamina. The lamina forms a molecular interface between the inner nuclear envelope membrane and chromatin. In lesser concentrations, the lamins are distributed throughout the nucleoplasm. In humans, there are two major types of lamins: A-type lamins (consisting of lamins A and C), found primarily in differentiated cells, and B-type lamins (lamins B1 and B2) that are found in all nucleated cells.
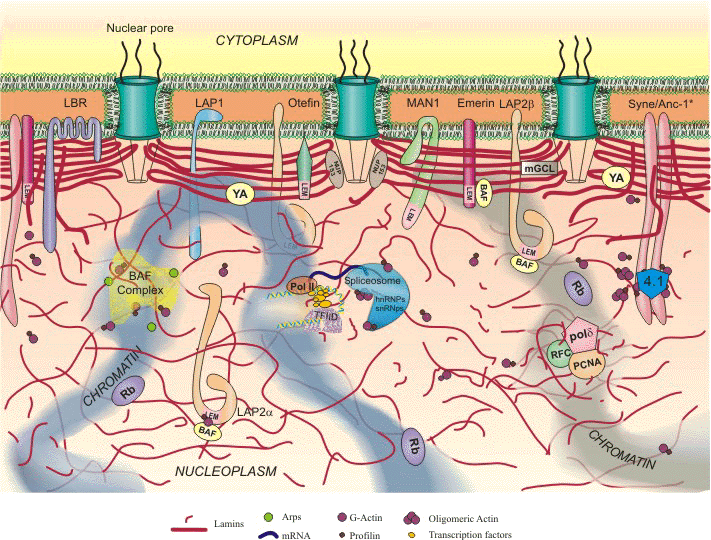
Lamins have a central rod domain resembling that of cytoskeletal IF proteins, with the exception that there are an additional six heptads in coil 1B. In vitro studies have shown that dimers are the basic building blocks of higher order lamin structures. However , little information is available regarding the precise types of structures lamins form within nuclei. In vitro, however, lamin dimers interact in a head-to-tail fashion to form protofilaments. These protofilaments subsequently aggregate to form paracrystalline arrays. Most lamins are not capable of assembling into 10nm diameter IF in vitro.
Function
Nuclear lamins are involved in a number of essential nuclear functions, including nuclear envelope assembly and disassembly during cell division, DNA synthesis, transcription, and apoptosis. Nuclear lamins have been found to co-localize with DNA synthesis sites. Studies have shown that the addition of dominant-negative mutant lamins to Xenopus nuclei results in co-localization of PCNA and factor C (RFC), two known replication factors, with the lamin aggregates. With the breakdown of the lamina, it was also shown that inhibition of DNA synthesis >95%, implying that lamins are integral to DNA synthesis. Lamins are also believed to regulate transcription. LA and LC bind to transcription factors such as pRB, which binds to LAP2 . Disruption of lamin assembly results in significant inhibition of pol II activity and alteration in the splitting factors B” and Y12’s formations.
Diseases
Many interesting possibilities for lamin functions are being revealed by the recent discoveries showing that a remarkable array of diseases is linked to mutations in human lamin A. To date lamin mutations are responsible for a wide array of diseases. These include Emery–Dreifuss muscular dystrophy (EDMD); dilated cardiomyopathy (DCM); familial partial lipodystrophy (FPLD); mandibuloacral dysplasia (MAD); autosomal recessive Charcot–Marie–Tooth disorder type 2 (AR-CMT2); limb girdle muscular dystrophy 1B (LGMD-1B) and the premature aging diseases Hutchinson-Gilford Progeria Syndrome (HGPS) and atypical forms of Werners Disease . The age of onset and the symptoms of these diseases vary over a wide range, but there are overlapping similarities, including muscle wasting, locomotory problems, fat redistribution, and cardiovascular problems. The nuclei in cells derived from patients with these “laminopathies” are frequently abnormal in shape and numerous components of these diseased nuclei are altered including nuclear pore complexes.
The type V intermediate filament proteins are the nuclear lamins. These are considered to be nucleoskeletal IF proteins that are determinants of nuclear size, shape, mechanical integrity, and positioning of nuclear pores. They also play important roles in DNA replication, transcription and the disassembly of the nucleus in mitosis and the reassembly of the nucleus in daughter cells.
Lamins are are concentrated in the nuclear lamina. The lamina forms a molecular interface between the inner nuclear envelope membrane and chromatin. In lesser concentrations, the lamins are distributed throughout the nucleoplasm. In humans, there are two major types of lamins: A-type lamins (consisting of lamins A and C), found primarily in differentiated cells, and B-type lamins (lamins B1 and B2) that are found in all nucleated cells.
Lamins have a central rod domain resembling that of cytoskeletal IF proteins, with the exception that there are an additional six heptads in coil 1B. In vitro studies have shown that dimers are the basic building blocks of higher order lamin structures. However , little information is available regarding the precise types of structures lamins form within nuclei. In vitro, however, lamin dimers interact in a head-to-tail fashion to form protofilaments. These protofilaments subsequently aggregate to form paracrystalline arrays. Most lamins are not capable of assembling into 10nm diameter IF in vitro.
Function
Nuclear lamins are involved in a number of essential nuclear functions, including nuclear envelope assembly and disassembly during cell division, DNA synthesis, transcription, and apoptosis. Nuclear lamins have been found to co-localize with DNA synthesis sites. Studies have shown that the addition of dominant-negative mutant lamins to Xenopus nuclei results in co-localization of PCNA and factor C (RFC), two known replication factors, with the lamin aggregates. With the breakdown of the lamina, it was also shown that inhibition of DNA synthesis >95%, implying that lamins are integral to DNA synthesis. Lamins are also believed to regulate transcription. LA and LC bind to transcription factors such as pRB, which binds to LAP2 . Disruption of lamin assembly results in significant inhibition of pol II activity and alteration in the splitting factors B” and Y12’s formations.
Diseases
Many interesting possibilities for lamin functions are being revealed by the recent discoveries showing that a remarkable array of diseases is linked to mutations in human lamin A. To date lamin mutations are responsible for a wide array of diseases. These include Emery–Dreifuss muscular dystrophy (EDMD); dilated cardiomyopathy (DCM); familial partial lipodystrophy (FPLD); mandibuloacral dysplasia (MAD); autosomal recessive Charcot–Marie–Tooth disorder type 2 (AR-CMT2); limb girdle muscular dystrophy 1B (LGMD-1B) and the premature aging diseases Hutchinson-Gilford Progeria Syndrome (HGPS) and atypical forms of Werners Disease . The age of onset and the symptoms of these diseases vary over a wide range, but there are overlapping similarities, including muscle wasting, locomotory problems, fat redistribution, and cardiovascular problems. The nuclei in cells derived from patients with these “laminopathies” are frequently abnormal in shape and numerous components of these diseased nuclei are altered including nuclear pore complexes.