


Peroxisomes
http://reasonandscience.heavenforum.org/t2162-peroxisomes
Summary Peroxisomes are specialized for carrying out oxidation reactions using molecular oxygen. They generate hydrogen peroxide, which they employ for oxidative purposes— and contain catalase to destroy the excess. Like mitochondria and plastids, peroxisomes are self-replicating organelles. Because they do not contain DNA or ribosomes, however, all of their proteins are encoded in the cell nucleus. Some of these proteins are conveyed to peroxisomes via peroxisomal precursor vesicles that bud from the ER, but most are synthesized in the cytosol and directly imported. A specific sequence of three amino acids near the C-terminus of many of the latter proteins functions as a peroxisomal import signal. The mechanism of protein import differs from that of mitochondria and chloroplasts, in that even oligomeric proteins are imported from the cytosol without unfolding.
Peroxisomes differ from mitochondria and chloroplasts in many ways. Most notably, they are surrounded by only a single membrane, and they do not contain DNA or ribosomes. Thus, because peroxisomes lack a genome, all of their proteins are encoded in the nucleus. Peroxisomes acquire most of these proteins by selective import from the cytosol, although some of them enter the peroxisome membrane via the ER.
Like mitochondria, peroxisomes are major sites of oxygen utilization. One hypothesis is that peroxisomes are a vestige of an ancient organelle that performed all the oxygen metabolism in the primitive ancestors of eukaryotic cells. When the oxygen produced by photosynthetic bacteria first accumulated in the atmosphere, it would have been highly toxic to most cells. Peroxisomes might have lowered the intracellular concentration of oxygen, while also exploiting its chemical reactivity to perform useful oxidation reactions. According to this view, the later development of mitochondria rendered peroxisomes largely obsolete because many of the same biochemical reactions—which had formerly been carried out in peroxisomes without producing energy—were now coupled to ATP formation by means of oxidative phosphorylation. The oxidation reactions performed by peroxisomes in present-day cells could therefore partly be those whose functions were not taken over by mitochondria.
Peroxisomes Use Molecular Oxygen and Hydrogen Peroxide to Perform Oxidation Reactions
Peroxisomes are so named because they usually contain one or more enzymes that use molecular oxygen to remove hydrogen atoms from specific organic substrates (designated here as R) in an oxidation reaction that produces hydrogen peroxide (H2O2):
RH2 + O2 → R + H2O2
Catalase uses the H2O2 generated by other enzymes in the organelle to oxidize a variety of other substrates—including formic acid, formaldehyde, and alcohol—by the “peroxidation” reaction: H2O2 + R′H2 → R′ + 2H2O. This type of oxidation reaction is particularly important in liver and kidney cells, where the peroxisomes detoxify various harmful molecules that enter the bloodstream. About 25% of the ethanol we drink is oxidized to acetaldehyde in this way. In addition, when excess H2O2 accumulates in the cell, catalase converts it to H2O through the reaction 2H2O2 → 2H2O + O2. A major function of the oxidation reactions performed in peroxisomes is the breakdown of fatty acid molecules. The process, called β oxidation, shortens the alkyl chains of fatty acids sequentially in blocks of two carbon atoms at a time, thereby converting the fatty acids to acetyl CoA. The peroxisomes then export the acetyl CoA Acetyl-CoAto the cytosol for use in biosynthetic reactions. In mammalian cells, β oxidation occurs in both mitochondria and peroxisomes; in yeast and plant cells, however, this essential reaction occurs exclusively in peroxisomes. An essential biosynthetic function of animal peroxisomes is to catalyze the first reactions in the formation of plasmalogens, which are the most abundant class of phospholipids in myelin
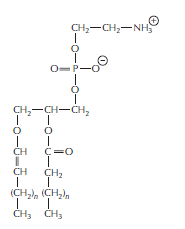
The structure of a plasmalogen. Plasmalogens are very abundant in the myelin sheaths that insulate the axons of nerve cells. They make up some 80–90% of the myelin membrane phospholipids. In addition to an ethanolamine head group and a longchain fatty acid attached to the same glycerol phosphate backbone used for phospholipids, plasmalogens contain an unusual fatty alcohol that is attached through an ether linkage (bottom left).
Plasmalogen deficiencies cause profound abnormalities in the myelination of nerve-cell axons, which is one reason why many peroxisomal disorders lead to neurological disease. Peroxisomes are unusually diverse organelles, and even in the various cell types of a single organism they may contain different sets of enzymes. They also adapt remarkably to changing conditions. Yeasts grown on sugar, for example, have few small peroxisomes. But when some yeasts are grown on methanol, numerous large peroxisomes are formed that oxidize methanol; and when grown on fatty acids, they develop numerous large peroxisomes that break down fatty acids to acetyl CoA by β oxidation. Peroxisomes are also important in plants. Two types of plant peroxisomes have been studied extensively. One is present in leaves, where it participates in photorespiration (Figure A below).
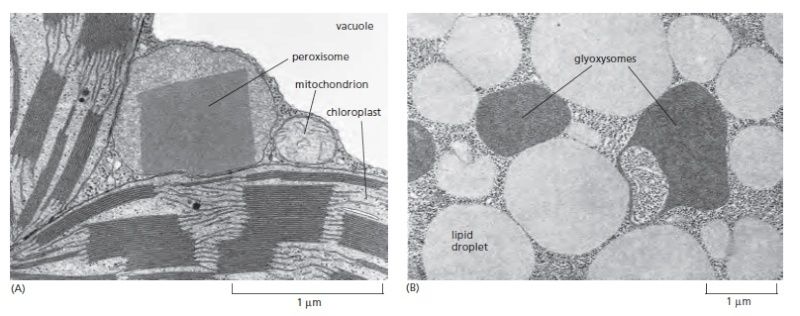
Electron micrographs of two types of peroxisomes found in plant cells. (A) A peroxisome with a paracrystalline core in a tobacco leaf mesophyll cell. Its close association with chloroplasts is thought to facilitate the exchange of materials between these organelles during photorespiration. The vacuole in plant cells is equivalent to the lysosome in animal cells. (B) Peroxisomes in a fat-storing cotyledon cell of a tomato seed 4 days after germination. Here the peroxisomes (glyoxysomes) are associated with the lipid droplets that store fat, reflecting their central role in fat mobilization and gluconeogenesis during seed germination.
The other type of peroxisome is present in germinating seeds, where it converts the fatty acids stored in seed lipids into the sugars needed for the growth of the young plant. Because this conversion of fats to sugars is accomplished by a series of reactions known as the glyoxylate cycle, these peroxisomes are also called glyoxysomes (Figure B above).
In the glyoxylate cycle, two molecules of acetyl CoA produced by fatty acid breakdown in the peroxisome are used to make succinic acid, which then leaves the peroxisome and is converted into glucose in the cytosol. The glyoxylate cycle does not occur in animal cells, and animals are therefore unable to convert the fatty acids in fats into carbohydrates.
A Short Signal Sequence Directs the Import of Proteins into Peroxisomes
A specific sequence of three amino acids (Ser–Lys–Leu) located at the C-terminus of many peroxisomal proteins functions as an import signal See table below:
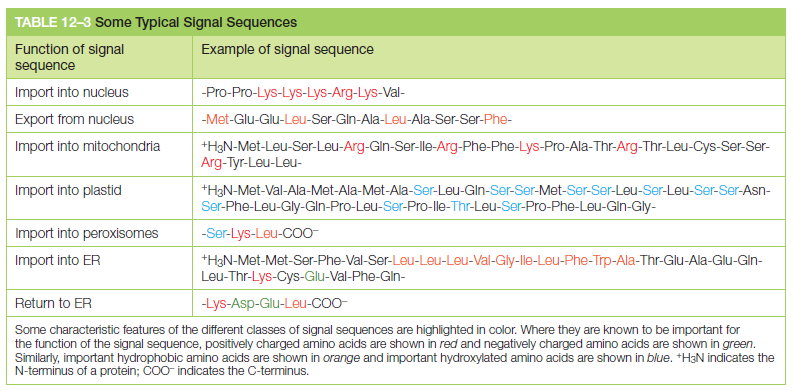
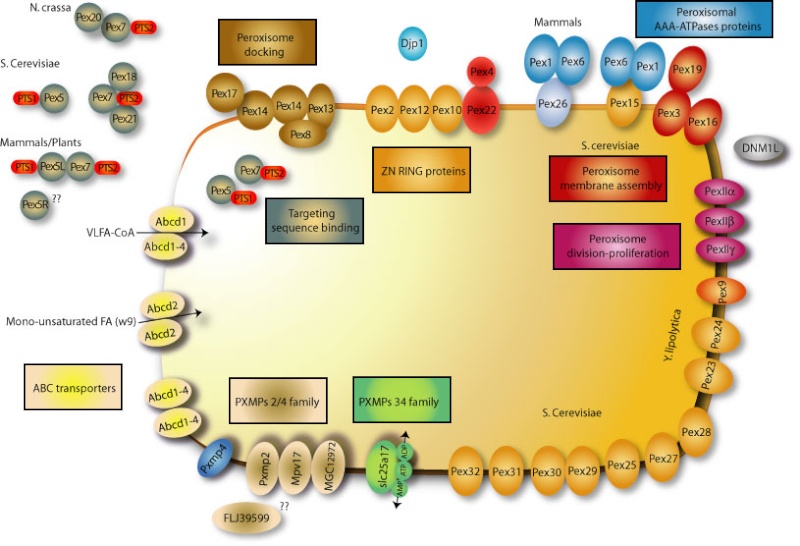
Other peroxisomal proteins contain a signal sequence near the N-terminus. If either sequence is attached to a cytosolic protein, the protein is imported into peroxisomes. The import signals are first recognized by soluble receptor proteins in the cytosol. Numerous distinct proteins, called peroxins, participate in the import process, which is driven by ATP hydrolysis. A complex of at least six different peroxins forms a protein translocator in the peroxisome membrane.
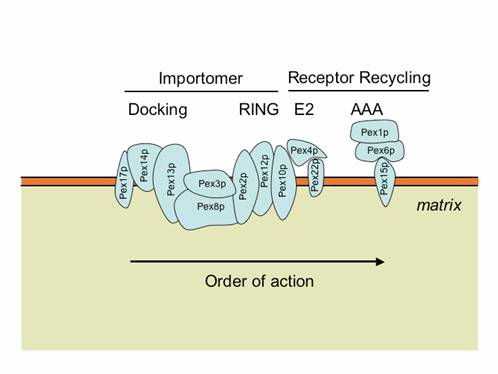
The importomer plays a role in matrix, but not membrane, protein import.5 The PTS1 and PTS2 receptors and their accessory proteins (e.g. Pex20p) ferry cargo from the cytosol and first interact with the docking subcomplex . The receptor/cargo complexes then either enter the matrix, or are deeply embedded in the peroxisome membrane. This is followed by cargo release into the peroxisome matrix, export/release of the receptors on the peroxisome membrane , followed by dislocation/recycling of the receptors from a peroxisome-associated state to the cytosol. Mutations in any component of the importomer affect the import of peroxisomal matrix proteins, suggesting that the whole importomer is somehow involved in protein translocation across this membrane . However, certain transient residents of the peroxisome matrix, such as Pex5p and Pex20p, become peroxisome-associated and protease protected, even in the absence of the RING subcomplex of the importomer, but their entry into the peroxisome is Pex14p-dependent. These data suggest that the docking subcomplex may be the true translocon, at least for these proteins, if not for other matrix cargoes as well. The RING subcomplex proteins are required for the export/release of receptors on the peroxisome membrane. The dislocation/recycling of the receptors from the peroxisomes to the cytosol requires the action of a receptor recycling machinery, comprised of an E2-like ubiquitin-conjugating enzyme, Pex4p, two AAA ATPases, Pex1p and Pex6p, that interact with each other in an ATP-dependent manner, and a peroxisomal membrane protein (Pex15p in S. cerevisiae or PEX26 in mammals), which provides a docking site for Pex6p . When this receptor recycling machinery is affected, a peroxisomal RADAR (acronym for Receptor Accumulation and Degradation in the Absence of Recycling) pathway becomes evident . This involves polyubiquitylation of peroxisome-membrane-associated Pex5p and Pex20p, most likely by redundant UBCs (Ubc1/Ubc4p/Ubc5p in S. cerevisiae), followed by their degradation by proteasomes.
Even oligomeric proteins do not have to unfold to be imported. To allow the passage of such compactly folded cargo molecules, the pore formed by the transporter is thought to be dynamic in its dimensions, adapting in size to the particular cargo molecules to be transported. In this respect, the mechanism differs from that used by mitochondria and chloroplasts. One soluble import receptor, the peroxin Pex5 recognizes the C-terminal peroxisomal import signal. It accompanies its cargo all the way into peroxisomes and, after cargo release, cycles back to the cytosol. After delivering its cargo to the peroxisome lumen, Pex5 undergoes ubiquitilation. This modification is required to release Pex5 back into the cytosol, where the ubiquitin is removed. An ATPase composed of Pex1 and Pex6 harnesses the energy of ATP hydrolysis to help release Pex5 from peroxisomes. The importance of this import process and of peroxisomes is demonstrated by the inherited human disease Zellweger syndrome, in which a defect in importing proteins into peroxisomes leads to a profound peroxisomal deficiency. These individuals, whose cells contain “empty” peroxisomes, have severe abnormalities in their brain, liver, and kidneys, and they die soon after birth. A mutation in the gene encoding peroxin Pex5 causes one form of the disease. A defect in Pex7, the receptor for the N-terminal import signal, causes a milder peroxisomal disease. It has long been debated whether new peroxisomes arise from preexisting ones by organelle growth and fission—like for mitochondria and plastids— or whether they derive as a specialized compartment from the endoplasmic reticulum (ER). Aspects of both views are true
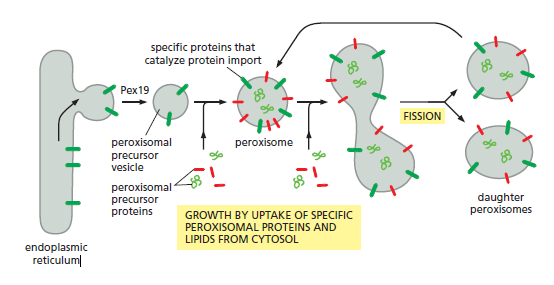
A model that explains how peroxisomes proliferate and how new peroxisomes arise. Peroxisomal precursor vesicles bud from the ER. At least two peroxisomal membrane proteins, Pex3 and Pex15, follow this route. The machinery that drives the budding reaction and that selects only peroxisomal proteins for packaging into these vesicles depends on Pex19 and other cytosolic proteins that are still unknown. Peroxisomal precursor vesicles may then fuse with one another or with preexisting peroxisomes. The peroxisome membrane contains import receptors and protein translocators that are required for the import of peroxisomal proteins made on cytosolic ribosomes, including new copies of the import receptors and translocator components. Presumably, the lipids required for growth are also imported, although some may derive directly from the ER in the membrane of peroxisomal precursor vesicles.
Most peroxisomal membrane proteins are made in the cytosol and insert into the membrane of preexisting peroxisomes, but others are first integrated into the ER membrane,
where they are packaged into specialized peroxisomal precursor vesicles. New precursor vesicles may then fuse with one another and begin importing additional peroxisomal proteins, using their own protein import machinery to grow into mature peroxisomes, which can undergo cycles of growth and fission.
Peroxisomes are highly adaptable organelles that carry out oxidative reactions. Distinct cellular machineries act together to coordinate peroxisome formation, growth, division, inheritance, turnover, movement and function. Soluble and membrane-associated components of these machineries form complex networks of physical and functional interactions that provide supramolecular control of the precise dynamics of peroxisome biogenesis. 4
In eukaryotes, lipid metabolism requires the function of peroxisomes. These multitasking organelles are also part of species-specific pathways such as the glyoxylate cycle in yeast and plants or the synthesis of ether lipid in mammals. Proteins required for the biogenesis of peroxisomes typically assemble in large molecular complexes, which participate in membrane formation, protein transport, peroxisome duplication and - inheritance during cell division. Peroxisomal function is essential for life. Mutations in PEX genes, encoding for biogenesis factors, are often associated with lethal disorders. The association of peroxisomes with other organelles suggests an extensive participation in organellar crosstalk. 1
Protein Import into Peroxisomes 2
Abstract
Peroxisomes are essential intracellular organelles that involve many metabolic processes, such as β-oxidation of very long-chain fatty acids and synthesis of plasmalogen and bile acids as well as generation and degradation of Hydrogen peroxide. These peroxisomal functions are fulfilled by strictly and spatiotemporally regulated compartmentation of the proteins catalysing these reactions. Defects in peroxisomal protein import results in inherited peroxisome biogenesis disorders in humans. Peroxisomal matrix and membrane proteins are synthesised on free ribosomes but transported into peroxisomes by distinct pathways determined by specific targeting signals and their receptors. The mechanism by which this is achieved has been clarified by identification of many PEX genes and the products named peroxins, the essential factors for peroxisome biogenesis.
 3
3
 1
1
Key Concepts
Peroxisome plays an essential role in various metabolic pathways.
Deficiency of peroxisomes in human causes severe foetal genetic diseases, peroxisome-deficient disorders.
Functions and the integrity of peroxisomes are archived by proper import of both matrix and membrane proteins into peroxisomes.
Peroxisomal proteins are encoded by
-nuclear DNA,
-synthesised in cytosolic free ribosomes and
-post-translationally imported into pre-existing peroxisomes.
Peroxisomal matrix and membrane proteins are translocated into peroxisomes via different pathways in a manner dependent on their distinct targeting signals.
Many of peroxins encoded by PEX genes are responsible factors for peroxisome biogenesis and are involved in the import of peroxisomal proteins.
Various experimental approaches have elucidated the molecular mechanisms underlying peroxisome biogenesis.
Semi-permeabilised cells, in which only the plasma membrane is selectively permeabilised, are used to investigate peroxisomal protein import, as with living cells.
1) http://www.springer.com/us/book/9783709117873
2) http://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0002618.pub2/abstract
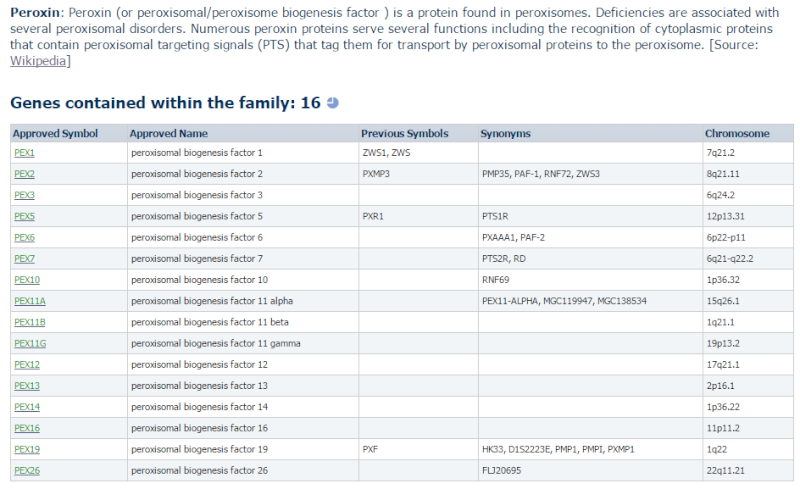
3) http://www.genenames.org/cgi-bin/genefamilies/set/957
4) http://www.nature.com/nrm/journal/v2/n5/full/nrm0501_357a.html
5) http://labs.biology.ucsd.edu/subramani/research.htm
http://reasonandscience.heavenforum.org/t2162-peroxisomes
Summary Peroxisomes are specialized for carrying out oxidation reactions using molecular oxygen. They generate hydrogen peroxide, which they employ for oxidative purposes— and contain catalase to destroy the excess. Like mitochondria and plastids, peroxisomes are self-replicating organelles. Because they do not contain DNA or ribosomes, however, all of their proteins are encoded in the cell nucleus. Some of these proteins are conveyed to peroxisomes via peroxisomal precursor vesicles that bud from the ER, but most are synthesized in the cytosol and directly imported. A specific sequence of three amino acids near the C-terminus of many of the latter proteins functions as a peroxisomal import signal. The mechanism of protein import differs from that of mitochondria and chloroplasts, in that even oligomeric proteins are imported from the cytosol without unfolding.
Peroxisomes differ from mitochondria and chloroplasts in many ways. Most notably, they are surrounded by only a single membrane, and they do not contain DNA or ribosomes. Thus, because peroxisomes lack a genome, all of their proteins are encoded in the nucleus. Peroxisomes acquire most of these proteins by selective import from the cytosol, although some of them enter the peroxisome membrane via the ER.
Like mitochondria, peroxisomes are major sites of oxygen utilization. One hypothesis is that peroxisomes are a vestige of an ancient organelle that performed all the oxygen metabolism in the primitive ancestors of eukaryotic cells. When the oxygen produced by photosynthetic bacteria first accumulated in the atmosphere, it would have been highly toxic to most cells. Peroxisomes might have lowered the intracellular concentration of oxygen, while also exploiting its chemical reactivity to perform useful oxidation reactions. According to this view, the later development of mitochondria rendered peroxisomes largely obsolete because many of the same biochemical reactions—which had formerly been carried out in peroxisomes without producing energy—were now coupled to ATP formation by means of oxidative phosphorylation. The oxidation reactions performed by peroxisomes in present-day cells could therefore partly be those whose functions were not taken over by mitochondria.
Peroxisomes Use Molecular Oxygen and Hydrogen Peroxide to Perform Oxidation Reactions
Peroxisomes are so named because they usually contain one or more enzymes that use molecular oxygen to remove hydrogen atoms from specific organic substrates (designated here as R) in an oxidation reaction that produces hydrogen peroxide (H2O2):
RH2 + O2 → R + H2O2
Catalase uses the H2O2 generated by other enzymes in the organelle to oxidize a variety of other substrates—including formic acid, formaldehyde, and alcohol—by the “peroxidation” reaction: H2O2 + R′H2 → R′ + 2H2O. This type of oxidation reaction is particularly important in liver and kidney cells, where the peroxisomes detoxify various harmful molecules that enter the bloodstream. About 25% of the ethanol we drink is oxidized to acetaldehyde in this way. In addition, when excess H2O2 accumulates in the cell, catalase converts it to H2O through the reaction 2H2O2 → 2H2O + O2. A major function of the oxidation reactions performed in peroxisomes is the breakdown of fatty acid molecules. The process, called β oxidation, shortens the alkyl chains of fatty acids sequentially in blocks of two carbon atoms at a time, thereby converting the fatty acids to acetyl CoA. The peroxisomes then export the acetyl CoA Acetyl-CoAto the cytosol for use in biosynthetic reactions. In mammalian cells, β oxidation occurs in both mitochondria and peroxisomes; in yeast and plant cells, however, this essential reaction occurs exclusively in peroxisomes. An essential biosynthetic function of animal peroxisomes is to catalyze the first reactions in the formation of plasmalogens, which are the most abundant class of phospholipids in myelin
The structure of a plasmalogen. Plasmalogens are very abundant in the myelin sheaths that insulate the axons of nerve cells. They make up some 80–90% of the myelin membrane phospholipids. In addition to an ethanolamine head group and a longchain fatty acid attached to the same glycerol phosphate backbone used for phospholipids, plasmalogens contain an unusual fatty alcohol that is attached through an ether linkage (bottom left).
Plasmalogen deficiencies cause profound abnormalities in the myelination of nerve-cell axons, which is one reason why many peroxisomal disorders lead to neurological disease. Peroxisomes are unusually diverse organelles, and even in the various cell types of a single organism they may contain different sets of enzymes. They also adapt remarkably to changing conditions. Yeasts grown on sugar, for example, have few small peroxisomes. But when some yeasts are grown on methanol, numerous large peroxisomes are formed that oxidize methanol; and when grown on fatty acids, they develop numerous large peroxisomes that break down fatty acids to acetyl CoA by β oxidation. Peroxisomes are also important in plants. Two types of plant peroxisomes have been studied extensively. One is present in leaves, where it participates in photorespiration (Figure A below).
Electron micrographs of two types of peroxisomes found in plant cells. (A) A peroxisome with a paracrystalline core in a tobacco leaf mesophyll cell. Its close association with chloroplasts is thought to facilitate the exchange of materials between these organelles during photorespiration. The vacuole in plant cells is equivalent to the lysosome in animal cells. (B) Peroxisomes in a fat-storing cotyledon cell of a tomato seed 4 days after germination. Here the peroxisomes (glyoxysomes) are associated with the lipid droplets that store fat, reflecting their central role in fat mobilization and gluconeogenesis during seed germination.
The other type of peroxisome is present in germinating seeds, where it converts the fatty acids stored in seed lipids into the sugars needed for the growth of the young plant. Because this conversion of fats to sugars is accomplished by a series of reactions known as the glyoxylate cycle, these peroxisomes are also called glyoxysomes (Figure B above).
In the glyoxylate cycle, two molecules of acetyl CoA produced by fatty acid breakdown in the peroxisome are used to make succinic acid, which then leaves the peroxisome and is converted into glucose in the cytosol. The glyoxylate cycle does not occur in animal cells, and animals are therefore unable to convert the fatty acids in fats into carbohydrates.
A Short Signal Sequence Directs the Import of Proteins into Peroxisomes
A specific sequence of three amino acids (Ser–Lys–Leu) located at the C-terminus of many peroxisomal proteins functions as an import signal See table below:
Other peroxisomal proteins contain a signal sequence near the N-terminus. If either sequence is attached to a cytosolic protein, the protein is imported into peroxisomes. The import signals are first recognized by soluble receptor proteins in the cytosol. Numerous distinct proteins, called peroxins, participate in the import process, which is driven by ATP hydrolysis. A complex of at least six different peroxins forms a protein translocator in the peroxisome membrane.
The importomer plays a role in matrix, but not membrane, protein import.5 The PTS1 and PTS2 receptors and their accessory proteins (e.g. Pex20p) ferry cargo from the cytosol and first interact with the docking subcomplex . The receptor/cargo complexes then either enter the matrix, or are deeply embedded in the peroxisome membrane. This is followed by cargo release into the peroxisome matrix, export/release of the receptors on the peroxisome membrane , followed by dislocation/recycling of the receptors from a peroxisome-associated state to the cytosol. Mutations in any component of the importomer affect the import of peroxisomal matrix proteins, suggesting that the whole importomer is somehow involved in protein translocation across this membrane . However, certain transient residents of the peroxisome matrix, such as Pex5p and Pex20p, become peroxisome-associated and protease protected, even in the absence of the RING subcomplex of the importomer, but their entry into the peroxisome is Pex14p-dependent. These data suggest that the docking subcomplex may be the true translocon, at least for these proteins, if not for other matrix cargoes as well. The RING subcomplex proteins are required for the export/release of receptors on the peroxisome membrane. The dislocation/recycling of the receptors from the peroxisomes to the cytosol requires the action of a receptor recycling machinery, comprised of an E2-like ubiquitin-conjugating enzyme, Pex4p, two AAA ATPases, Pex1p and Pex6p, that interact with each other in an ATP-dependent manner, and a peroxisomal membrane protein (Pex15p in S. cerevisiae or PEX26 in mammals), which provides a docking site for Pex6p . When this receptor recycling machinery is affected, a peroxisomal RADAR (acronym for Receptor Accumulation and Degradation in the Absence of Recycling) pathway becomes evident . This involves polyubiquitylation of peroxisome-membrane-associated Pex5p and Pex20p, most likely by redundant UBCs (Ubc1/Ubc4p/Ubc5p in S. cerevisiae), followed by their degradation by proteasomes.
Even oligomeric proteins do not have to unfold to be imported. To allow the passage of such compactly folded cargo molecules, the pore formed by the transporter is thought to be dynamic in its dimensions, adapting in size to the particular cargo molecules to be transported. In this respect, the mechanism differs from that used by mitochondria and chloroplasts. One soluble import receptor, the peroxin Pex5 recognizes the C-terminal peroxisomal import signal. It accompanies its cargo all the way into peroxisomes and, after cargo release, cycles back to the cytosol. After delivering its cargo to the peroxisome lumen, Pex5 undergoes ubiquitilation. This modification is required to release Pex5 back into the cytosol, where the ubiquitin is removed. An ATPase composed of Pex1 and Pex6 harnesses the energy of ATP hydrolysis to help release Pex5 from peroxisomes. The importance of this import process and of peroxisomes is demonstrated by the inherited human disease Zellweger syndrome, in which a defect in importing proteins into peroxisomes leads to a profound peroxisomal deficiency. These individuals, whose cells contain “empty” peroxisomes, have severe abnormalities in their brain, liver, and kidneys, and they die soon after birth. A mutation in the gene encoding peroxin Pex5 causes one form of the disease. A defect in Pex7, the receptor for the N-terminal import signal, causes a milder peroxisomal disease. It has long been debated whether new peroxisomes arise from preexisting ones by organelle growth and fission—like for mitochondria and plastids— or whether they derive as a specialized compartment from the endoplasmic reticulum (ER). Aspects of both views are true
A model that explains how peroxisomes proliferate and how new peroxisomes arise. Peroxisomal precursor vesicles bud from the ER. At least two peroxisomal membrane proteins, Pex3 and Pex15, follow this route. The machinery that drives the budding reaction and that selects only peroxisomal proteins for packaging into these vesicles depends on Pex19 and other cytosolic proteins that are still unknown. Peroxisomal precursor vesicles may then fuse with one another or with preexisting peroxisomes. The peroxisome membrane contains import receptors and protein translocators that are required for the import of peroxisomal proteins made on cytosolic ribosomes, including new copies of the import receptors and translocator components. Presumably, the lipids required for growth are also imported, although some may derive directly from the ER in the membrane of peroxisomal precursor vesicles.
Most peroxisomal membrane proteins are made in the cytosol and insert into the membrane of preexisting peroxisomes, but others are first integrated into the ER membrane,
where they are packaged into specialized peroxisomal precursor vesicles. New precursor vesicles may then fuse with one another and begin importing additional peroxisomal proteins, using their own protein import machinery to grow into mature peroxisomes, which can undergo cycles of growth and fission.
Peroxisomes are highly adaptable organelles that carry out oxidative reactions. Distinct cellular machineries act together to coordinate peroxisome formation, growth, division, inheritance, turnover, movement and function. Soluble and membrane-associated components of these machineries form complex networks of physical and functional interactions that provide supramolecular control of the precise dynamics of peroxisome biogenesis. 4
In eukaryotes, lipid metabolism requires the function of peroxisomes. These multitasking organelles are also part of species-specific pathways such as the glyoxylate cycle in yeast and plants or the synthesis of ether lipid in mammals. Proteins required for the biogenesis of peroxisomes typically assemble in large molecular complexes, which participate in membrane formation, protein transport, peroxisome duplication and - inheritance during cell division. Peroxisomal function is essential for life. Mutations in PEX genes, encoding for biogenesis factors, are often associated with lethal disorders. The association of peroxisomes with other organelles suggests an extensive participation in organellar crosstalk. 1
Protein Import into Peroxisomes 2
Abstract
Peroxisomes are essential intracellular organelles that involve many metabolic processes, such as β-oxidation of very long-chain fatty acids and synthesis of plasmalogen and bile acids as well as generation and degradation of Hydrogen peroxide. These peroxisomal functions are fulfilled by strictly and spatiotemporally regulated compartmentation of the proteins catalysing these reactions. Defects in peroxisomal protein import results in inherited peroxisome biogenesis disorders in humans. Peroxisomal matrix and membrane proteins are synthesised on free ribosomes but transported into peroxisomes by distinct pathways determined by specific targeting signals and their receptors. The mechanism by which this is achieved has been clarified by identification of many PEX genes and the products named peroxins, the essential factors for peroxisome biogenesis.
3 1Key Concepts
Peroxisome plays an essential role in various metabolic pathways.
Deficiency of peroxisomes in human causes severe foetal genetic diseases, peroxisome-deficient disorders.
Functions and the integrity of peroxisomes are archived by proper import of both matrix and membrane proteins into peroxisomes.
Peroxisomal proteins are encoded by
-nuclear DNA,
-synthesised in cytosolic free ribosomes and
-post-translationally imported into pre-existing peroxisomes.
Peroxisomal matrix and membrane proteins are translocated into peroxisomes via different pathways in a manner dependent on their distinct targeting signals.
Many of peroxins encoded by PEX genes are responsible factors for peroxisome biogenesis and are involved in the import of peroxisomal proteins.
Various experimental approaches have elucidated the molecular mechanisms underlying peroxisome biogenesis.
Semi-permeabilised cells, in which only the plasma membrane is selectively permeabilised, are used to investigate peroxisomal protein import, as with living cells.
1) http://www.springer.com/us/book/9783709117873
2) http://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0002618.pub2/abstract
3) http://www.genenames.org/cgi-bin/genefamilies/set/957
4) http://www.nature.com/nrm/journal/v2/n5/full/nrm0501_357a.html
5) http://labs.biology.ucsd.edu/subramani/research.htm
Last edited by Admin on Sat Nov 18, 2017 10:16 am; edited 4 times in total