

Origin of CRISPR-Cas molecular complexes of prokaryotes
https://reasonandscience.catsboard.com/t3243-origin-of-crispr-cas-molecular-complexes-of-prokaryotes
1. All entrance identity check systems based on data collection and storage mechanisms are designed.
2. CRISPR-Cas is an immune system based on data storage and identity check systems.
3. Therefore, it was designed.
Casey Luskin: Researchers Suggest Molecular Machine Is Irreducibly Complex August 8, 2014,
https://evolutionnews.org/2014/08/researchers_sug/
Structure of molecular machine that targets viral DNA for destruction determined
"The structure of this biological machine is conceptually similar to an engineer's blueprint, and it explains how each of the parts in this complex assemble into a functional complex that efficiently identifies viral DNA when it enters the cell," Wiedenheft said. "This surveillance machine consists of 12 different parts and each part of the machine has a distinct job. If we're missing one part of the machine, it doesn't work."
https://www.sciencedaily.com/releases/2014/08/140807154143.htm
R.N. JACKSON: Crystal structure of the CRISPR RNA–guided surveillance complex from Escherichia coli 7 Aug 2014
https://www.science.org/doi/10.1126/science.1256328
Programmable Memory in Prokaryotes April 12, 2017
https://evolutionnews.org/2017/04/programmable-memory-in-prokaryotes/
Luciano A. Marraffini (2018): Many bacteria and archaea have the unique ability to heritably alter their genomes by incorporating small fragments of foreign DNA, called spacers, into CRISPR loci. Once transcribed and processed into individual CRISPR RNAs, spacer sequences guide Cas effector nucleases to destroy complementary, invading nucleic acids. Collectively, these two processes are known as the CRISPR–Cas immune response. 25
Udi Qimron: CRISPR and their associated proteins comprise a significant prokaryotic defense system against viruses and horizontally transferred nucleic acids 26
https://www.youtube.com/watch?v=qc6xgb4VXl0
CRISPR is actually a sequence of information that is present in the bacterial genome and it is regularly interspersed and repetitive in nature that's why it's called palindromic repeats and there is a protein that is associated with CRISPR locus which is known as CRISPR associated protein line which works like a molecular scissor. CRISPR can be understood as a defense mechanism against viruses that are used against phage viruses so the phage viruses are the key enemies of e.coli which infect them and kill them so the virus has developed a defense mechanism that is embedded in their genome and which is in terms of a DNA sequence CRISPR locus and a cas gene which is producing a protein cas9. The sequence information of the CRISPR locus is strikingly similar to the sequence information present in the phage genome scientists scratch their heads why this sequence information is identical and similar what is the consequence of that or what is the cause of that? The phage infected the bacteria and the fed genetic material is inside the bacteria and tries to get integrated into the bacterial genome these sequences remain incorporated in the CRISPR locus now once the sequence gets incorporated into the CRISPR locus they become a part of the bacterial genome. Let's say there is a second infection by the same patch now this CRISPR locus would create specific RNA which is targeting the phage genome and the cas gene product which is cas9 would get associated with that RNA which is produced by the CRISPR locus called guide RNA and another RNA called tracer RNA and ultimately this cas9 along with this tracer and guide RNA cleaves the fudge genome now once the phage genome is cleaved using this CRISPR cas9 system the phage DNA would be degraded and that is how the bacteria protects itself from phage viruses and that's how it works it's a bacterial adaptive immune system so precise and beautiful.
Pascale Cossart (2016): In nature, bacteria need to defend themselves constantly, particularly against bacteriophages (or phages), the viruses that specifically attack bacteria. A phage generally attaches itself to a bacterium, injects its DNA into it, and subverts the bacterium’s mechanisms of replication, transcription, and translation in order to replicate itself. The phage DNA reproduces its own DNA, transcribes it into RNA, and produces phage proteins that accumulate to generate new phages and eventually cause the bacterial cell to explode (or lyse), releasing hundreds of new bacteriophages. Phages continually infect bacteria everywhere—in soil, in water, and even in our own intestinal microbiota. Bacteriophage families are numerous and vary widely in their form, size, composition, and the bacteria they target. To begin their attack, bacteriophages need a site of attachment, a particular component on the surface of a bacterium. This site of attachment is specific for each virus and the bacteria that it can infect. Bacteria have an immune system called CRISPR. CRISPR regions in the chromosomes allow bacteria to recognize predators, particularly previously encountered phages, and to destroy them. CRISPR regions protect and essentially “vaccinate” bacteria against bacteriophages. In fact, it has been shown that bacteria can be artificially vaccinated! When a population of bacteria is inoculated with a phage, a small number survive and are able to integrate a fragment of the phage DNA into their genome, in the region called the CRISPR locus. This allows the bacteria, if the phage ever attacks again, to recognize the phage DNA and degrade it. This ingenious phenomenon, known as interference, occurs due to the structure of the CRISPR region and to cas genes (CRISPR-associated genes) located near this region. The CRISPR locus is a region of the chromosome composed of repeated sequences of around 50 nucleotides, interspersed with sequences known as spacers that are similar to those of bacteriophages. Some bacteria have several CRISPR loci with different sequence repetitions. Around 40% of bacteria have one or more CRISPRs, whereas others have none. CRISPR loci can be quite long, sometimes with more than 100 repetitions and spacers. CRISPRs have two functions: acquisition and interference. Acquisition, also called adaptation, is the process of acquiring fragments of DNA from a phage, and interference is the immunization process by Cas proteins encoded by cas genes (Fig. below).
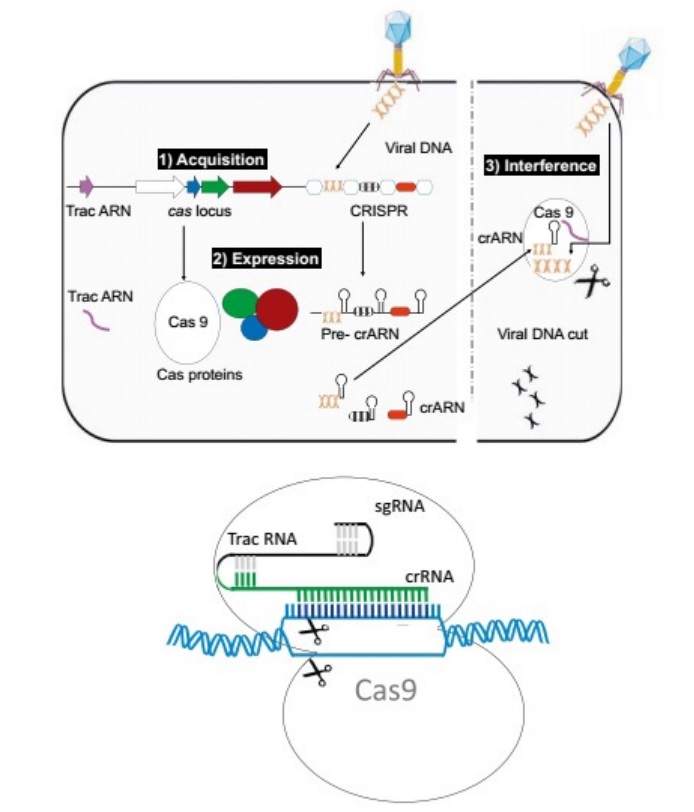
(Top) The three steps involved in CRISPR function.
(1) Integration of a piece of DNA from a phage into the CRISPR locus (acquisition);
(2) the expression of Cas proteins and of pre-crRNA, which is then split into small crRNAs;
(3) the interference that takes place when the DNA injected by a phage into a bacterium meets a crRNA, a hybrid form that is then degraded, consequently preventing infection.
(Bottom) Schematic drawing of genome modification (gene editing) by an sgRNA (small guided RNA) made of crRNA and tracrRNA and the endonuclease Cas9.
Bacteria have numerous proteins with various complementary and synergistic functions in the process of adaptation and interference. They permit the addition of DNA fragments into the CRISPR locus, but their main purpose is to react to invading phages. The CRISPR locus is transcribed into a long CRISPR RNA, which is then split into small RNAs called crRNAs, each containing a spacer and a part of the repeated sequence. When a phage injects its DNA into the bacterium, the crRNA recognizes and binds to it. An enzyme then recognizes the hybrid and cleaves the phage DNA at the point where the crRNA has paired. Replication of the phage DNA is inactivated, and the infection is stopped. Genome editing or modification is the identification of the proteins involved in the cleavage of the hybrid DNA. This process is performed by a complex of proteins containing the protein Cas1 and sometimes by a single protein called Cas9. Cas9 is unique in that it can attach itself to a DNA strand and, due to the two distinct domains of its structure, cut this DNA on each of its two strands. This protein is the basis of the CRISPR/Cas9 technology, which enables a variety of genome modifications and mutations in mammals, plants, insects, and fish in addition to bacteria. This system works due to the Cas9 protein and also a guide RNA hybrid that is made from one RNA similar to the region to be mutated and a second RNA called tracrRNA, or trans-activating crRNA. tracrRNA was discovered next to the CRISPR locus in Streptococcus pyogenes and was shown to be homologous to the repeated regions of the locus, enabling it to guide the Cas9 protein and the crRNA toward the target. In summary, by expressing the Cas9 protein with a composite RNA made up of an identical sequence to the target region, a tracrRNA, and a complementary fragment to the tracrRNA, one can now introduce a mutation or deletion into a target genome of any origin. After the 2012 publication in Science of the elegant studies by the teams led by Emmanuelle Charpentier and Jennifer Doudna, the CRISPR method was so intriguing that it provoked an avalanche of research and publications demonstrating that this technique could be used in many cases and with many variations. 24
The CRISPR–Cas system
Tina Y.Liu (2020): CRISPR-Cas systems stand out as the only known RNA programmed pathways for detecting and destroying bacteriophages and plasmids. . Class 1 CRISPR-Cas systems, the most widespread and diverse of these adaptive immune systems, use an RNA-guided multi-protein complex to find foreign nucleic acids and trigger their destruction. These multisubunit complexes target and cleave DNA and RNA, and regulatory molecules control their activities. CRISPR-Cas loci constitute the only known adaptive immune system in bacteria and archaea. They typically include an array of repeat sequences (CRISPRs) with intervening “spacers” matching sequences of DNA or RNA from viruses or other mobile genetic elements, and a set of genes encoding CRISPR-associated (Cas) proteins.
Transcription across the CRISPR array produces a precursor crRNA (pre-crRNA) that is processed by nucleases into small, non-coding CRISPR RNAs (crRNAs). Each crRNA molecule assembles with one or more Cas proteins into an effector complex that binds crRNA-complementary regions in foreign DNA or RNA. The effector complex then triggers degradation of the targeted DNA or RNA using either an intrinsic nuclease activity or a separate nuclease in trans.
Giedrius Gasiunas (2012):The silencing of invading nucleic acids is executed by ribonucleoprotein complexes preloaded with small, interfering CRISPR RNAs (crRNAs) that act as guides for targeting and degradation of foreign nucleic acid. The Cas9–crRNA complex of the Streptococcus thermophilus CRISPR3/Cas system introduces a double-strand break at a specific site in DNA containing a sequence complementary to crRNA. DNA cleavage is executed by Cas9, which uses two distinct active sites, RuvC and HNH, to generate site-specific nicks on opposite DNA strands. Results demonstrate that the Cas9–crRNA complex functions as an RNA-guided endonuclease with RNA-directed target sequence recognition and protein-mediated DNA cleavage. 20
J.Cepelewicz (2020): CRISPR acts like an adaptive immune system; it enables bacteria that have been exposed to a virus to pass on a genetic “memory” of that infection to their descendants, which can then mount better defenses against a repeat infection. It’s a system that works so well that an estimated half of all bacterial species use CRISPR. Researchers have uncovered dozens of other systems that bacteria use to rebuff phage invasions. But in laboratory studies, bacteria primarily develop what’s known as surface-based phage resistance. Mutations change receptor molecules on the surface of the bacterial cell, so that the phage can no longer recognize and invade it.
The strategy is akin to shutting a door and throwing away the key: It offers the bacteria complete safety from infection by the virus. But that protection comes at a significant price, because it also disrupts whatever nutrient uptake, waste disposal, communication task or other cellular function the receptor would have been providing — taking a constant toll on a cell’s fitness.
In contrast, CRISPR only drags on a cell’s resources when it’s active, during a viral infection. Even so, CRISPR represents a riskier gambit: It doesn’t start to work until phages have already entered the cell, meaning that there’s a chance the viruses could overcome it. And CRISPR doesn’t just attack viral DNA; it can also prevent bacteria from taking up beneficial genes from other microbes, like those that confer antibiotic resistance. What factors affect the trade-offs in costs and fitness? For the past six years, Edze Westra, an evolutionary ecologist at the University of Exeter in England, has led a team pursuing the answer to that question. In 2015, they discovered that nutrient availability and phage density affected whether Pseudomonas bacteria relied on surface-based or CRISPR-based resistance. In environments poor in resources, receptor modifications were more burdensome, so CRISPR became a better bargain. When resources were plentiful, bacteria grew more densely and phage epidemics became more frequent. Bacteria then faced greater selective pressure to close themselves off from infection entirely, and so they shut down receptors to gain surface-based resistance. This explained why surface-based resistance was so common in laboratory cultures. Growing in a test tube rich in nutrients, “these bacteria are on a holiday,” Westra said. “They are having a terrific time.”
Still, these rules weren’t cut and dried. Plenty of bacteria in natural high-nutrient environments use CRISPR, and plenty of bacteria in natural low-nutrient environments don’t. “It’s all over the place,” Westra said. “That told us that we were probably still missing something.”
How Biodiversity Reshapes the Battle
Then one of Westra’s graduate students, Ellinor Opsal, proposed another potential factor: the diversity of the biological communities in which bacteria live. This factor is harder to study, but scientists had previously observed that it could affect phage immunity in bacteria. For example, in 2005, James Bull, a biologist at the University of Texas, Austin, and William Harcombe, his graduate student at the time (now at the University of Minnesota), found that E. coli bacteria didn’t evolve immunity to a phage when a second bacterial species was present. Similarly, Britt Koskella, an evolutionary biologist at the University of California, Berkeley, and one of her graduate students, Catherine Hernandez, reported last year that phage resistance failed to arise in Pseudomonas bacteria living on their actual host (a plant), though they always gained immunity in a test tube. Could the diversity of the surroundings influence not just whether or not resistance to phages evolved, but the nature of that resistance?
To find out, Westra’s team performed a new set of experiments: Instead of altering the nutrient conditions for Pseudomonas bacteria growing with phages, they added three other bacterial species — species that competed against Pseudomonas for resources but weren’t targeted by the phage. Left to themselves, Pseudomonas would normally develop surface-based mutations. But in the company of rivals, they were far more likely to turn to CRISPR. Further investigation showed that the more complex community dynamics had shifted the fitness costs: The bacteria could no longer afford to inactivate receptors because they not only had to survive the phage, but also had to outcompete the bacteria around them. These results from Westra’s group dovetail with earlier findings that phages can produce greater diversity in bacterial communities. “Now, that diversity is actually feeding back to the phage side of things” by affecting phage resistance, Koskella said. “It’s neat to see that coming full circle.” By understanding that kind of feedback loop, she added, “we can start to ask more general questions about the impacts that phages have in a community context.”
For one, the bacteria’s shift toward a CRISPR-based phage response had another, broader effect. When Westra’s group grew Pseudomonas in moth larvae hosts, they found that the bacteria with surface-based resistance were less virulent, killing the larvae much more slowly than the bacteria with active CRISPR systems did. 18
Discovering CRISPR
S.H. Sternberg (2015): The CRISPR locus was first identified in Escherichia coli as an unusual series of 29-bp repeats separated by 32-bp spacer sequences (Ishino et al., 1987) 21 Carl Zimmer tells us the story (2015): The scientists who discovered CRISPR had no way of knowing that they had discovered something so revolutionary. They didn’t even understand what they had found. In 1987, Yoshizumi Ishino and colleagues at Osaka University in Japan published the sequence of a gene called iap belonging to the gut microbe E. coli. To better understand how the gene worked, the scientists also sequenced some of the DNA surrounding it. They hoped to find spots where proteins landed, turning iap on and off. But instead of a switch, the scientists found something incomprehensible. Near the iap gene lay five identical segments of DNA. DNA is made up of building blocks called bases, and the five segments were each composed of the same 29 bases. These repeat sequences were separated from each other by 32-base blocks of DNA, called spacers. Unlike the repeat sequences, each of the spacers had a unique sequence.
This peculiar genetic sandwich didn’t look like anything biologists had found before. When the Japanese researchers published their results, they could only shrug. “The biological significance of these sequences is not known,” they wrote. It was hard to know at the time if the sequences were unique to E. coli, because microbiologists only had crude techniques for deciphering DNA. But in the 1990s, technological advances allowed them to speed up their sequencing. By the end of the decade, microbiologists could scoop up seawater or soil and quickly sequence much of the DNA in the sample. This technique — called metagenomics — revealed those strange genetic sandwiches in a staggering number of species of microbes. They became so common that scientists needed a name to talk about them, even if they still didn’t know what the sequences were for. In 2002, Ruud Jansen of Utrecht University in the Netherlands and colleagues dubbed these sandwiches “clustered regularly interspaced short palindromic repeats” — CRISPR for short.
Jansen’s team noticed something else about CRISPR sequences: They were always accompanied by a collection of genes nearby. They called these genes Cas genes, for CRISPR-associated genes. The genes encoded enzymes that could cut DNA, but no one could say why they did so, or why they always sat next to the CRISPR sequence. Three years later, three teams of scientists independently noticed something odd about CRISPR spacers. They looked a lot like the DNA of viruses. “And then the whole thing clicked,” said Eugene Koonin. At the time, Koonin, an evolutionary biologist at the National Center for Biotechnology Information in Bethesda, Md., had been puzzling over CRISPR and Cas genes for a few years. As soon as he learned of the discovery of bits of virus DNA in CRISPR spacers, he realized that microbes were using CRISPR as a weapon against viruses.
Koonin knew that microbes are not passive victims of virus attacks. They have several lines of defense. Koonin thought that CRISPR and Cas enzymes provide one more. In Koonin’s hypothesis, bacteria use Cas enzymes to grab fragments of viral DNA. They then insert the virus fragments into their own CRISPR sequences. Later, when another virus comes along, the bacteria can use the CRISPR sequence as a cheat sheet to recognize the invader.
Scientists didn’t know enough about the function of CRISPR and Cas enzymes for Koonin to make a detailed hypothesis. But his thinking was provocative enough for a microbiologist named Rodolphe Barrangou to test it. To Barrangou, Koonin’s idea was not just fascinating, but potentially a huge deal for his employer at the time, the yogurt maker Danisco. Danisco depended on bacteria to convert milk into yogurt, and sometimes entire cultures would be lost to outbreaks of bacteria-killing viruses. Now Koonin was suggesting that bacteria could use CRISPR as a weapon against these enemies.
To test Koonin’s hypothesis, Barrangou and his colleagues infected the milk-fermenting microbe Streptococcus thermophilus with two strains of viruses. The viruses killed many of the bacteria, but some survived. When those resistant bacteria multiplied, their descendants turned out to be resistant too. Some genetic change had occurred. Barrangou and his colleagues found that the bacteria had stuffed DNA fragments from the two viruses into their spacers. When the scientists chopped out the new spacers, the bacteria lost their resistance. Barrangou, now an associate professor at North Carolina State University, said that this discovery led many manufacturers to select for customized CRISPR sequences in their cultures, so that the bacteria could withstand virus outbreaks. “If you’ve eaten yogurt or cheese, chances are you’ve eaten CRISPR-ized cells,” he said.
In 2007, Blake Wiedenheft joined Doudna’s lab as a postdoctoral researcher, eager to study the structure of Cas enzymes to understand how they worked. Doudna agreed to the plan — not because she thought CRISPR had any practical value, but just because she thought the chemistry might be cool. “You’re not trying to get to a particular goal, except understanding,” she said. As Wiedenheft, Doudna and their colleagues figured out the structure of Cas enzymes, they began to see how the molecules worked together as a system. When a virus invades a microbe, the host cell grabs a little of the virus’s genetic material, cuts open its own DNA, and inserts the piece of virus DNA into a spacer. As the CRISPR region fills with virus DNA, it becomes a molecular most-wanted gallery, representing the enemies the microbe has encountered. The microbe can then use this viral DNA to turn Cas enzymes into precision-guided weapons. The microbe copies the genetic material in each spacer into an RNA molecule. Cas enzymes then take up one of the RNA molecules and cradle it. Together, the viral RNA and the Cas enzymes drift through the cell. If they encounter genetic material from a virus that matches the CRISPR RNA, the RNA latches on tightly. The Cas enzymes then chop the DNA in two, preventing the virus from replicating.
CRISPR, microbiologists realized, is also an adaptive immune system. It lets microbes learn the signatures of new viruses and remember them. And while we need a complex network of different cell types and signals to learn to recognize pathogens, a single-celled microbe has all the equipment necessary to learn the same lesson on its own. But how did microbes develop these abilities? Ever since microbiologists began discovering CRISPR-Cas systems in different species, Koonin and his colleagues have been reconstructing the systems’ evolution. CRISPR-Cas systems use a huge number of different enzymes, but all of them have one enzyme in common, called Cas1. The job of this universal enzyme is to grab incoming virus DNA and insert it in CRISPR spacers. Recently, Koonin and his colleagues discovered what may be the origin of Cas1 enzymes.
Along with their own genes, microbes carry stretches of DNA called mobile elements that act like parasites. The mobile elements contain genes for enzymes that exist solely to make new copies of their own DNA, cut open their host’s genome, and insert the new copy. Sometimes mobile elements can jump from one host to another, either by hitching a ride with a virus or by other means, and spread through their new host’s genome.
Koonin and his colleagues discovered that one group of mobile elements, called casposons, makes enzymes that are pretty much identical to Cas1. In a new paper in Nature Reviews Genetics, Koonin and Mart Krupovic of the Pasteur Institute in Paris argue that the CRISPR-Cas system got its start when mutations transformed casposons from enemies into friends. Their DNA-cutting enzymes became domesticated, taking on a new function: to store captured virus DNA as part of an immune defense. While CRISPR may have had a single origin, it has blossomed into a tremendous diversity of molecules. Koonin is convinced that viruses are responsible for this. Once they faced CRISPR’s powerful, precise defense, the viruses evolved evasions. Their genes changed sequence so that CRISPR couldn’t latch onto them easily. And the viruses also evolved molecules that could block the Cas enzymes. The microbes responded by evolving in their turn. They acquired new strategies for using CRISPR that the viruses couldn’t fight. Over many thousands of years, in other words, evolution behaved like a natural laboratory, coming up with new recipes for altering DNA. 17
Diversity, ecology, and evolution of the CRISPR-Cas systems
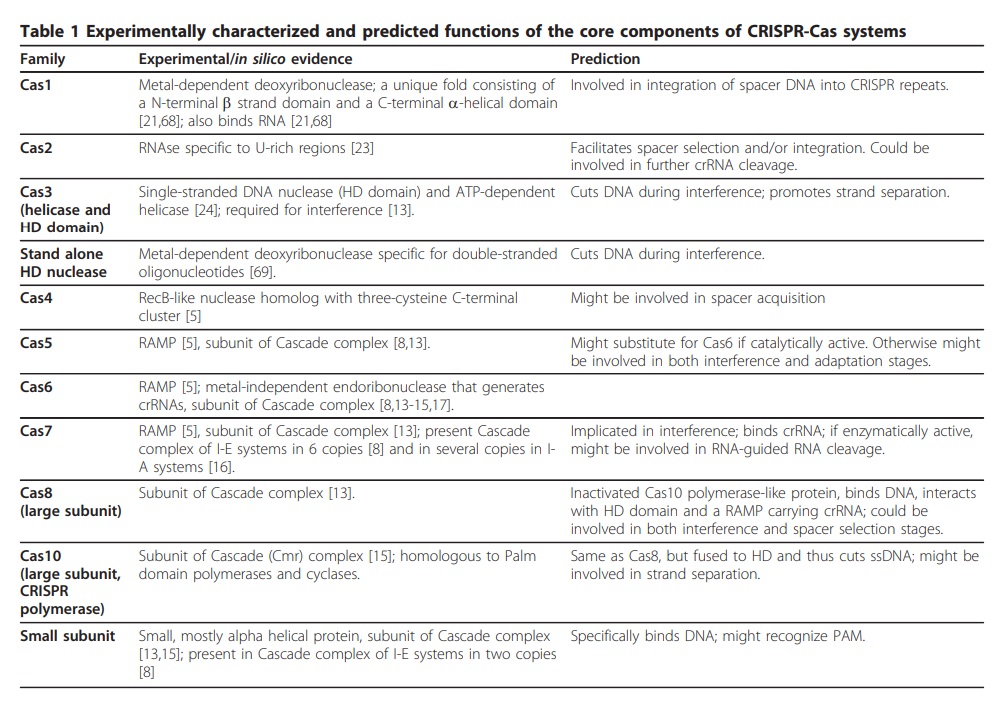
Devashish Rath (2015): The length and sequence of repeats and the length of spacers are well conserved within a CRISPR locus, but may vary between CRISPRs in the same or different genomes. Repeat sequences are in the range of 21 bp to 48 bp, and spacers are between 26 bp and 72 bp. The observed variation is perhaps not surprising given how widespread the system is. The number of spacers within a CRISPR locus varies widely; from a few to several hundred. Genomes can have single or multiple CRISPR loci and in some species, these loci can make up a significant part of the chromosome. Not all CRISPR loci have adjacent cas genes and instead rely on trans-encoded factors. (a trans-acting factor is usually a regulatory protein that binds to DNA). Another feature associated with CRISPR loci is the presence of a conserved sequence, called leader, located upstream of the CRISPR with respect to the direction of transcription. The Cas proteins are a highly diverse group. Many are predicted or identified to interact with nucleic acids; e.g. as nucleases, helicases and RNA-binding proteins. The Cas1 and Cas2 proteins are involved in adaptation and are virtually universal for CRISPR-Cas systems. Other Cas proteins are only associated with certain types of CRISPR-Cas systems. The diversity of Cas proteins, presence of multiple CRISPR loci, and frequent horizontal transfer of CRISPR-Cas systems make classification a complex task. The most adopted classification identifies Type I, II and III CRISPR-Cas systems, with each having several subgroups. Different types of CRISPR-Cas systems can co-exist in a single organism. Recently, a Type IV system was proposed, which contains several Cascade genes but no CRISPR, cas1 or cas2. Type IV complex would be guided by protein-DNA interaction, not by crRNA, and constitutes an innate immune system preset to attack certain sequences. The Type I systems are defined by the presence of the signature protein Cas3, a protein with both helicase and DNase domains responsible for degrading the target. Currently, six subtypes of the Type I system are identified (Type I-A through Type I-F) that have a variable number of cas genes. Apart from cas1, cas2 and cas3, all Type I systems encode a Cascade-like complex. Cascade binds crRNA and locates the target, and most variants are also responsible for processing the crRNA. Cascade also enhances spacer acquisition in some cases. In the Type I-A system, Cas3 is a part of the Cascade complex. The Type II CRISPR-Cas systems encode Cas1 and Cas2, the Cas9 signature protein, and sometimes a fourth protein (Csn2 or Cas4). Cas9 assists in adaptation participate in crRNA processing and cleave the target DNA assisted by crRNA and an additional RNA called tracrRNA. Type II systems have been divided into subtypes II-A and II-B but recently a third, II-C, has been suggested. The csn2 and cas4 genes, both encoding proteins involved in adaptation, are present in Type II-A and Type II-B, respectively, while Type II-C lacks a fourth gene. The Type III CRISPR-Cas systems contain the signature protein Cas10 with unclear function. Most Cas proteins are destined for the Csm (in Type III-A) or Cmr (in Type III-B) complexes, which are similar to Cascade. Interestingly, while all Type I and II systems are known to target DNA, Type III systems target DNA and/or RNA. So far, the Type II systems have been exclusively found in bacteria while the Type I and Type III systems occur both in bacteria and archaea. A large number of genomes with detected CRISPRs could be used as an argument for its importance as a defense mechanism. However, the CRISPR-Cas systems are probably mobile genetic elements that frequently transfer horizontally, which also contributes to their high prevalence. Other findings indicate that phages can still replicate in populations with one but not two spacers targeting them.
Eugene V. Koonin (2019): The number and diversity of known CRISPR–Cas systems have substantially increased in recent years. The new classification includes 2 classes, 6 types and 33 subtypes compared with 5 types and 16 subtypes in 2015. At the adaptation stage, a distinct complex of Cas proteins binds to a target DNA, often after recognizing a distinct, short motif known as a protospacer-adjacent motif (PAM), and cleaves out a portion of the target DNA, the protospacer. After duplication of the repeat at the 5ʹ end of the CRISPR array, the adaptation complex inserts the protospacer DNA into the array, so that it becomes a spacer. Some CRISPR–Cas systems employ an alternative mechanism of adaptation — namely, spacer acquisition from RNA, via reverse transcription by a reverse transcriptase encoded at the CRISPR–cas locus. At the expression stage, the CRISPR array is typically transcribed as a single transcript — the pre-CRISPR RNA (pre-crRNA) — that is processed into mature CRISPR RNAs (crRNAs), each containing the spacer sequence and parts of the flanking repeats. In different CRISPR–Cas variants, the pre-crRNA processing is mediated by a distinct subunit of a multiprotein Cas complex, by a single, multidomain Cas protein, or by non-Cas host RNases. At the interference stage, the crRNA, which typically remains bound to the processing complex (protein), serves as a guide to recognize the protospacer (or a closely similar sequence) in the invading genome of a virus or plasmid, which is then cleaved and inactivated by a Cas nuclease (or nucleases) that either is part of the effector or is recruited at the interference stage. The above summary is a brief, oversimplified description of the CRISPR–Cas functionality that inevitably omits many details. These can be found in recent reviews on different aspects of CRISPR–Cas biology. Similar to other biological defense mechanisms, archaeal and bacterial CRISPR–Cas systems show a remarkable diversity of Cas protein sequences, gene compositions, and architectures of the genomic loci. Our knowledge of this diversity is continuously expanding through the screening of ever-growing genomic and metagenomic databases. To keep pace with such expansion, a robust classification of CRISPR–Cas systems is essential for the progress of CRISPR research, but this presents formidable challenges, owing to the lack of universal markers and the fast evolution of the CRISPR–cas loci. Therefore, the two previous CRISPR–Cas classifications, published in Nature Reviews Microbiology in 2011 and 2015, employed a multipronged approach that combined comparisons of the gene compositions of CRISPR–Cas systems and their loci architectures with sequence similarity-based clustering and phylogenetic analysis of conserved Cas proteins, such as Cas1. The 2015 classification included 5 types and 16 subtypes, as well as introducing the major division of CRISPR–Cas systems into two classes that radically differ with respect to the architectures of their effector modules involved in crRNA processing and interference. The class 1 systems have effector modules composed of multiple Cas proteins, some of which form crRNA-binding complexes (such as the Cascade complex in type I systems) that, with contributions from additional Cas proteins, mediate pre-crRNA processing and interference. By contrast, class 2 systems encompass a single, multidomain crRNA-binding protein (such as Cas9 in type II systems) that combines all activities required for interference and, in some variants, also those involved in pre-crRNA processing (Box 1).
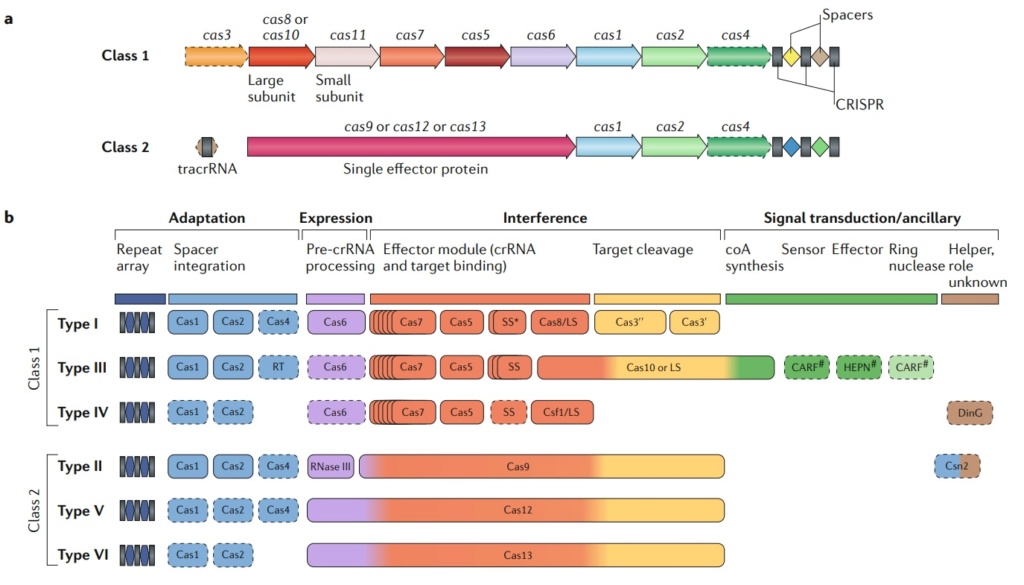
Class 1 CrisPr–Cas systems have effector modules composed of multiple Cas proteins that form a crrNa-binding complex and function together in binding and processing of the target. Class 2 systems have a single, multidomain. crrNa-binding protein that is functionally analogous to the entire effector complex of class 1. Part a of the figure illustrates the generic organizations of the class 1 and class 2 CrisPr–Cas loci. Part b of the figure shows the functional modules of CrisPr–Cas systems. the scheme shows the typical relationships between the genetic, structural and functional organizations of the six types of CrisPr–Cas systems. Protein names follow the current nomenclature. an asterisk indicates the putative small subunit that might be fused to the large subunit in several type i subtypes. the pound symbols (#) indicate that other unknown sensor, effector and ring nuclease protein families could be involved in the same signaling pathway. Dispensable (and/or missing, in some subtypes and variants) components are indicated by dashed outlines. Cas6 is shown with a thin solid outline for type i because it is dispensable in some, but not most, systems and with a dashed line for type iii because most of these systems apparently use the Cas6 protein provided in trans by other CrisPr–cas loci. the three colours for Cas9, Cas10, Cas12 and Cas13 reflect the fact that these proteins contribute to different stages of the CrisPr–Cas response. the CrisPr-associated rossmann fold (CarF) and higher eukaryotes and prokaryotes nucleotide-binding (HePN) domain proteins are the most common sensors and effectors, respectively, in the type III ancillary modules, but several alternative sensors and effectors have been identified, as well43. ring nucleases are a distinct variety of CarF domain proteins that cleave cyclic oligoa produced by Cas10 and thus control the indiscriminate rNase activity of the HePN domain of Csx1. Ls, large subunit; ss, small subunit; tracrrNa, transactivating CrisPr rNa.
CRISPR–Cas classification
No genes are shared by all CRISPR–Cas systems, ruling out the possibility of a straightforward, comprehensive phylogenetic classification analogous to that employed for cellular life forms.
Class 1 and its derivatives.
The classification of class 1 CRISPR–Cas systems, which include types I, III and IV, has remained relatively stable compared with the 2015 version (Fig. below).
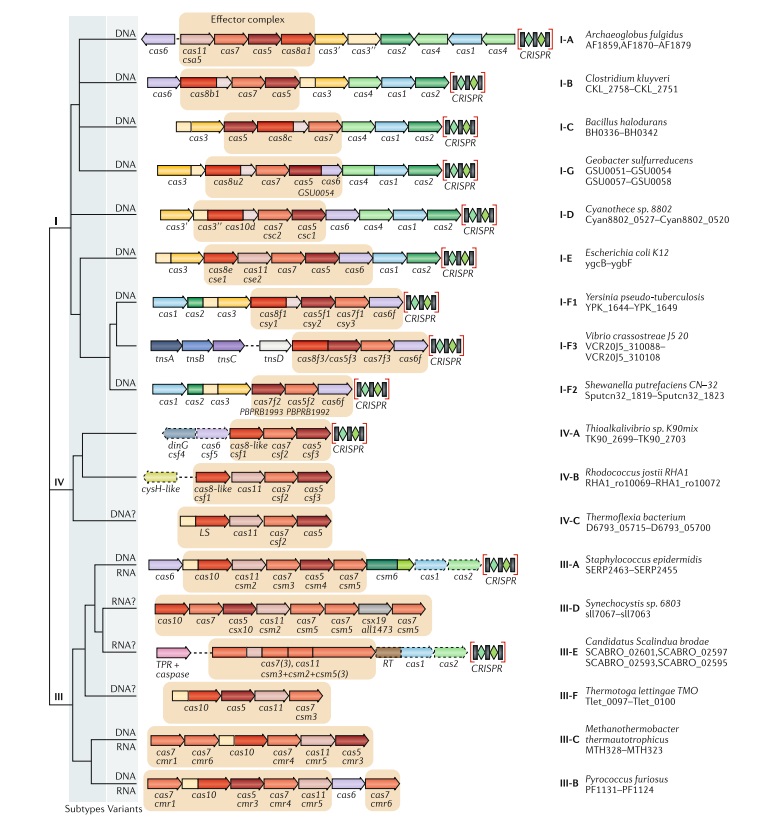
Updated classification of class 1 criSPr–cas systems.
The figure schematically shows representative (typical) CRISPR–cas loci of each class 1 subtype and of selected distinct variants, with the dendrogram on the left showing the likely evolutionary relationships between the types and subtypes. The column on the right indicates the organism and the corresponding gene range. Homologous genes are colour-coded and identified by a family name. The gene names follow the previous classification18. Where both a systematic name and a legacy name are commonly used, the legacy name is given under the systematic name. The small subunit is encoded by csm2, cmr5, cse2, csa5 and several additional families of homologous genes that are collectively denoted cas11. The adaptation module genes cas1 and cas2 are dispensable in subtypes III-A and III-E (dashed lines). Gene regions coloured cream represent the HD nuclease domain; the HD domain in Cas10 is distinct from that in Cas3 and Cas3ʹʹ. Functionally uncharacterized genes are shown in grey. The tan shading shows the effector module. The grey shading of different hues shows the two levels of classification: subtypes and variants. Most of the subtype III-B, III-C, III-E and III-F loci, as well as IV-B and IV-C loci, lack CRISPR arrays and are shown accordingly, although for each of the type III subtypes exceptions have been detected. CHAT, protease domain of the caspase family; RT, reverse transcriptase; TPR, tetratricopeptide repeat.
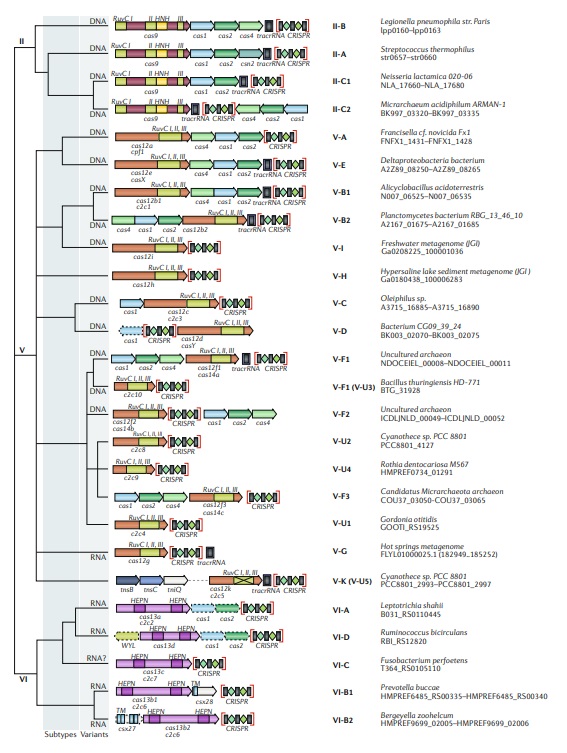
Updated classification of class 2 criSPr–cas systems.
The figure schematically shows representative (typical) CRISPR–cas loci for each class 2 subtype and for selected distinct variants, with the dendrogram on the left showing the likely evolutionary relationships between the types and subtypes. The column on the right indicates the organism and the corresponding gene range. Homologous genes are colour coded and are identified by a family name following the previous classification18. Where both a systematic name and a legacy name are commonly used, the legacy name is given under the systematic name. The grey shading of different hues shows the two levels of classification: subtypes and variants. The adaptation module genes cas1 and cas2 are present in only a subset of the subtype V-D, VI-A and VI-D loci and are accordingly shown by dashed lines. The WYL-domain-encoding genes and csx27 genes are also dispensable and shown by dashed lines. Additional genes encoding components of the interference module, such as transactivating CRISPR RNA (tracrRNA), are shown. The domains of the effector proteins are colour-coded: RuvC-like nuclease, green; HNH nuclease, yellow; higher eukaryotes and prokaryotes nucleotide-binding (HEPN) RNase, purple; transmembrane domains, blue.
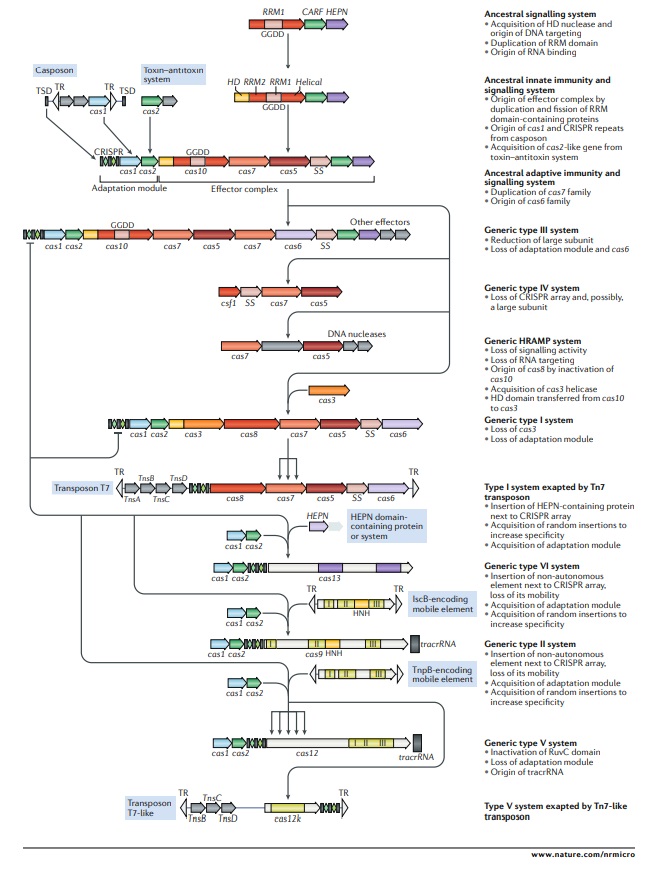
Outline of a complete scenario for the origins and evolution of criSPr–cas systems.
The figure depicts a hypothetical scenario of the origin of CRISPR–Cas systems from an ancestral signalling system (possibly an abortive infection defence system (Abi)). This putative ancestral Abi module shares a cyclic oligoA polymerase Palm domain (RNA recognition motif (RRM) fold) with Cas10 and is proposed to function analogously to type III CRISPR–Cas systems. Specifically, cyclic oligoA molecules that are synthesized in response to virus infection bind to the CRISPR-associated Rossmann fold (CARF) domain of the second protein in this system, resulting in activation of the RNase activity of the higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domain, which induces dormancy through indiscriminate RNA cleavage. This putative ancestral Abi module would give rise to the type III-like CRISPR–Cas effector module via duplication of the RRM domain, with subsequent inactivation of one of the copies (the two RRM domains are denoted RRM1 and RRM2). The ancestral class 1 CRISPR–Cas system is inferred to have evolved through the merger of two modules: the adaptation module, including the CRISPR repeats, derived from a casposon, and the type III-like effector module, likely derived from the ancestral Abi system. The subsequent acquisition of the HD nuclease domain by the effector module provided for RNA-guided DNA cleavage. Inactivation of the oligoA polymerase domain in the effector complex, or possibly replacement of Cas10 by an unrelated protein and acquisition of the Cas3 helicase, led to the emergence of type I systems, which lack the cyclic oligoA-dependent signalling pathway and exclusively cleave double-stranded DNA. Class 2 systems of type II and different subtypes of type V appear to have evolved independently by the recruitment of distinct TnpB nucleases that are encoded by IS605-like transposable elements. Type VI likely originated from an RNA-cleaving, HEPN domain-containing abortive infection or toxin–antitoxin system. Some CRISPR–Cas systems, such as type IV and Tn7-linked systems I-F3 and V-K, were subsequently recruited by mobile genetic elements and lost their interference capacity along with the original defence function. The key evolutionary events are described to the right of the images. The typical CRISPR–cas operon organization is shown for each CRISPR–Cas subtype and for selected distinct variants. Homologous genes are colour-coded and identified by a family name following the previous classification18. The multiforking arrows denote events that have been inferred to have occurred on multiple, independent occasions during the evolution of CRISPR–Cas systems. GGDD, key catalytic motif of the cyclase or polymerase domain of Cas10 that is involved in the synthesis of cyclic oligoA signalling molecules; HRAMP, haloarchaeal repeat-associated mysterious proteins; TR, terminal repeats; tracrRNA, transactivating CRISPR RNA; TSD, target site duplication, the likely source of ancestral repeats. 23
Imagine a company had the task to install a security system in its headquarters, based on biometrics. Biometrics comes from the Greek words “bios” (life) and “metrikos” (measure). It involves the implementation of a system that uses the analysis of biological characteristics of people, and that analyzes human characteristics for identity verification or identification. In order to distinguish employees that are permitted to enter the building, and exclude to enter those that are not welcome, there has to be first data collection and storage of the information in a memory bank. Every time, when someone arrives at the building, it will go through the security check, and the provided data will be compared to the data in the memory bank. If there is a match, the person is permitted to enter, or not.
Analogously, cells are capable of doing almost the same, with a few differences. They have an ingenious security check system, based on enemy recognition, and based on that knowledge, creating a sophisticated data bank, that is employed to recognize future enemy invasions, and annihilate them.
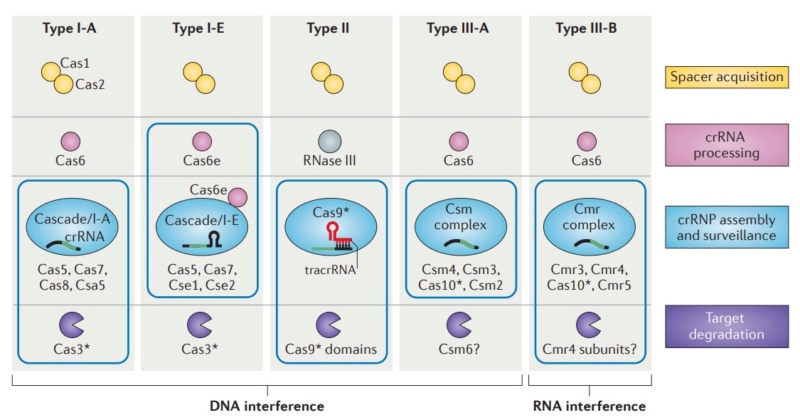
Diversity of CRISPR–Cas systems.
The CRISPR-associated (Cas) proteins can be divided into distinct functional categories as shown. The three types of CRISPR–Cas systems are defined on the basis of a type-specific signature Cas protein (indicated by an asterisk) and are further subdivided into subtypes. The CRISPR ribonucleoprotein (crRNP) complexes of type I and type III systems contain multiple Cas subunits, whereas the type II system contains a single Cas9 protein. Boxes indicate components of the crRNP complexes for each system. The type III-B system is unique in that it targets RNA, rather than DNA, for degradation.
Understanding CRISPR-Cas9
Eugene V Koonin (2011): CRISPR-Cas systems have three distinct functional stages of their operation. During the first stage, adaptation, short pieces of DNA (characteristic length of approximately 30 bp) homologous to virus or plasmid sequences (known as proto-spacers) are integrated into the CRISPR loci. The short (3 or 4 nucleotides) proto-spacer adjacent motifs (PAMs) located immediately downstream of the proto-spacer appear to determine the selection of the protospacer followed by integration into a pre-existing CRISPR array. The second stage, expression and processing, involves transcription and cleavage of long primary transcript of a CRISPR locus (pre-crRNA) that is processed into short crRNAs. This step is catalyzed by endoribonucleases encoded by the cas genes that either operate as a subunit of a larger complex (e.g. Cascade, CRISPR-associated complex for antiviral defense in Escherichia coli) or as a stand-alone enzyme, e.g., Cas6 in the archaeon Pyrococcus furiosus. At the third stage, interference, the alien nucleic acid (DNA or RNA) is targeted by a ribonucleoprotein complex containing a crRNA guide and a set of Cas proteins, and cleaved within or in the vicinity of the PAM sequence. In several CRISPR-Cas systems, crRNA have been shown to be complementary to either strand of the phage or plasmid which is best compatible with DNA being the target. Direct demonstration of DNA being the target of the CRISPR-Cas machinery has come from experiments in Staphylococcus epidermidis. In this case, insertion of a self-splicing intron into the proto-spacer sequence of the target gene rendered the respective plasmid resistant to the CRISPR-mediated immunity. 22
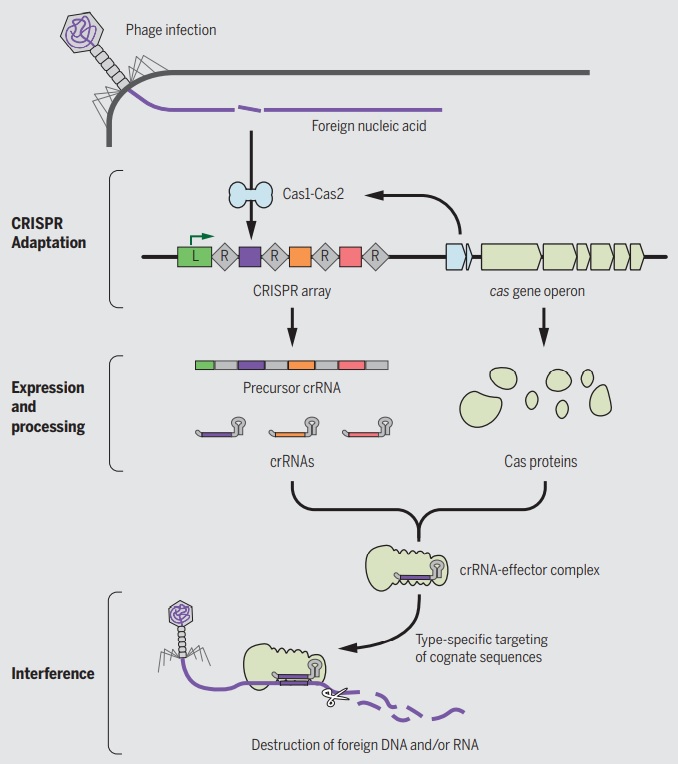
A roadmap of CRISPR-Cas adaptation and defense.
In the example illustrated, a bacterial cell is infected by a bacteriophage. The first stage of CRISPR-Cas defense is CRISPR adaptation. This involves the incorporation of small fragments of DNA from the invader into the host CRISPR array. This forms a genetic “memory” of the infection. The memories are stored as spacers (colored squares) between repeat sequences (R), and new spacers are added at the leader-proximal (L) end of the array. The Cas1 and Cas2 proteins, encoded within the cas gene operon, form a Cas1-Cas2 complex (blue)—the “workhorse” of CRISPR adaptation. In this example, the Cas1-Cas2 complex catalyzes the addition of a spacer from the phage genome (purple) into the CRISPR array. The second stage of CRISPR-Cas defense involves transcription of the CRISPR array and subsequent processing of the precursor transcript to generate CRISPR RNAs (crRNAs). Each crRNA contains a single spacer unit that is typically flanked by parts of the adjoining repeat sequences (gray). Individual crRNAs assemble with Cas effector proteins (light green) to form crRNA-effector complexes. The crRNA-effector complexes catalyze the sequence-specific recognition and destruction of foreign DNA and/or RNA elements. This process is known as interference. 13
 [/size]
[/size]
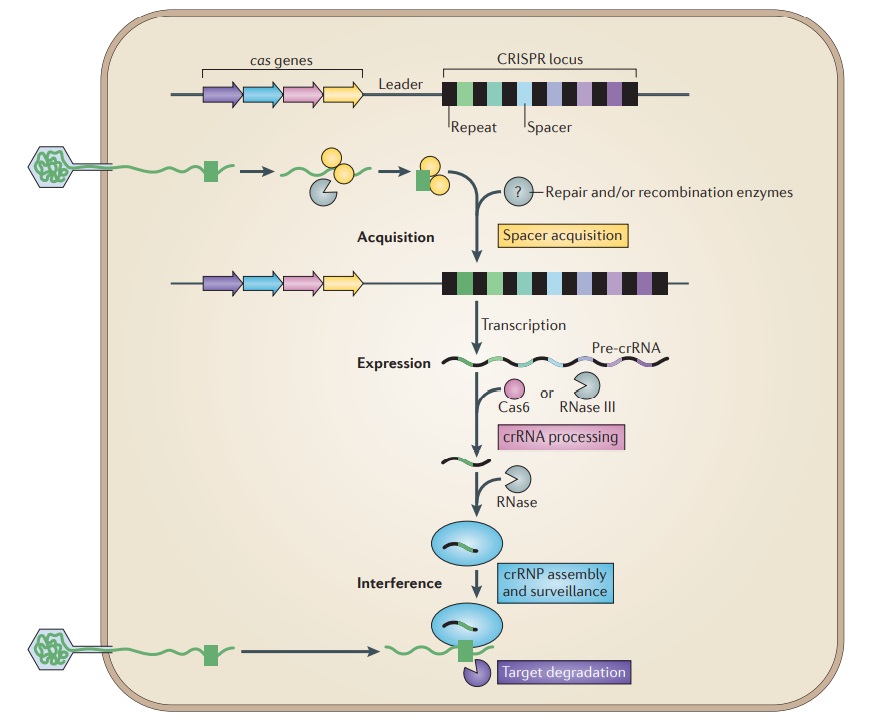
Overview of the CRISPR–Cas system.
Adaptive immunity by CRISPR–Cas systems is mediated by CRISPR RNAs (crRNAs) and Cas proteins, which form multicomponent CRISPR ribonucleoprotein (crRNP) complexes. The cas genes are colored according to function, as indicated by the four functional categories in coloured boxes:
spacer acquisition (yellow);
crRNA processing (pink);
crRNA assembly and surveillance (blue); and
target degradation (purple).
Involvement of non-Cas components (grey) is indicated, either when experimentally demonstrated (for example, RNase III processing in type II systems) or when anticipated (for example, the potential involvement of housekeeping repair and/or recombination enzymes). The first stage is known as acquisition, which occurs following the entry of an invading mobile genetic element (in this case, a viral genome). The invading DNA is fragmented and a new protospacer (green) is selected, processed and integrated as a new spacer at the leader end of the CRISPR array. During the second stage, which is known as expression, the CRISPR locus is transcribed and the pre-crRNA is processed into small crRNAs by CRISPR-associated (Cas6) and/or housekeeping ribonucleases (such as RNase III). The mature crRNAs and Cas proteins assemble to form a crRNP complex. During the final stage of interference, the crRNP scans invading DNA for a complementary nucleic acid target and on successful recognition, the target is eventually degraded by Cas nucleases. 27
S. H. Sternberg (2015): CRISPR-Cas immunity is conferred through integration of short DNA fragments into the CRISPR locus, and these spacer sequences record the history of past infections. The CRISPR locus is transcribed, and the resultant transcript is processed into shorter CRISPR-RNAs (crRNAs). CRISPR-Cas systems are classified as types I, II or III, which can be distinguished based on the presence of the signature Cas3, Cas9, or Cas10 genes, respectively. Type I are the most common, and much of our understanding of type I CRISPR-Cas systems comes from studies of E. coli Cascade (CRISPR-associated complex for antiviral defense), which is comprised of the five proteins Cse1, Cse2, Cas7, Cas5e, and Cas6e. These proteins assemble on a 61-nt crRNA, yielding a 405- kDa complex. The crRNA contains the 32-nt spacer sequence, which directs Cascade to sequences (protospacers) in foreign DNA, leading to formation of an R-loop intermediate. Cascade then recruits Cas3, which has an N-terminal histidine-aspartate (HD) nuclease domain and C-terminal superfamily 2 (SF2) helicase domain, to degrade the DNA. Cascade must discriminate between spacer sequences found in the bacterial chromosome and those found in foreign DNA. This discrimination is thought to be accomplished through recognition of a trinucleotide sequence motif called the protospacer-adjacent motif (PAM; 5′-A[A/T]G-3′ for E. coli Cascade), which is adjacent to the protospacer in foreign DNA, but absent in the CRISPR locus. Strict sequence requirements present a potential weakness because mutations in either the PAM or protospacer can allow foreign DNA to escape CRISPR-Cas immunity. However, bacteria can rapidly restore immunity using a positive-feedback loop to update the CRISPR locus. Priming requires Cascade with a crRNA bearing at least partial complementarity to the escape target, suggesting Cascade must be able to locate targets even when they bear mutations sufficient to escape immunity. Priming also requires Cas3 and the Cas1-Cas2 complex, which integrate new sequences into the CRISPR locus. The PAM-dependent pathway is highly efficient and allows Cascade to recruit Cas3 for strand-specific degradation of the target genome. The PAM-independent pathway is less efficient, but Cascade can still bind tightly to the DNA, ensuring that it can initiate the sequence of molecular events that precede primed spacer acquisition. Through this pathway, Cas3 recruitment becomes strictly dependent on Cas1-Cas2, and Cas1-Cas2 also attenuate Cas3 nuclease activity and enable Cas3 to rapidly translocate in either direction along the foreign DNA. These results establish Cas1-Cas2 as a trans-acting factor necessary for the recruitment and regulation of Cas3 at escape targets. Based on our findings, we propose a mechanistic framework describing how Cascade, Cas1, Cas2, and Cas3 work together to process and disable foreign genetic elements. 21
M. P. Terns et al. (2015): (CRISPR-Cas immune systems function to defend prokaryotes against potentially harmful mobile genetic elements including viruses and plasmids. The multiple CRISPR-Cas systems (Types I, II, and III) each target destruction of foreign nucleic acids via structurally and functionally diverse effector complexes (crRNPs). CRISPR-Cas effector complexes are comprised of CRISPR RNAs (crRNAs) that contain sequences homologous to the invading nucleic acids and Cas proteins specific to each immune system type. CRISPR-Cas systems confer prokaryotes with adaptive immunity against viruses, conjugative plasmids, and other potential genome invaders. A host CRISPR (clustered regularly interspaced short palindromic repeats) locus contains a leader region (typically 100–500 bp) followed by multiple copies of a repeat sequence (∼30–40 bp) separated by similarly sized, variable invader-derived sequences. Each crRNA contains a guide region comprised of invader-derived sequences that allow crRNA-Cas protein effector complexes to recognize and destroy invader nucleic acids. CRISPR-associated (Cas) proteins provide enzymatic machinery and structural components to carry out the distinct phases of the CRISPR-Cas pathway. Moreover, modules of Cas proteins (e.g., Csa, Cst, Cse, Csm, Cmr) comprise the distinct CRISPR-Cas immune systems: Type I (A-G), Type II (A-C), and Type III (A-B). 16
Dipali G Sashital (2019): Within this system, the CRISPR locus is programmed with ‘spacer’ sequences that are derived from foreign DNA and serve as a record of prior infection events 14 CRISPR cas9 in a bacteria acts as an adaptive immune response that is it remembers when a virus has infected the cell in the past and it keeps a little bit of viral DNA and stores it in a memory bank ( the spacers) and uses it so that if the same species of virus infects the cell again it will be able to compare the injected DNA to sequences in the data bank, recognize and respond to it quickly and effectively, and destroy it.
Devashish Rath (2015): The CRISPR-Cas mediated defense process can be divided into three stages.
1. Adaptation or spacer acquisition,where a short fragment of invading DNA is inserted into the CRISPR locus for future recognition of that invader;
2. crRNA biogenesis ( expression), which involves the biogenesis of guide RNA units (crRNA)
3. Target interference where these effector complexes vigilantly scan for and degrade invading genetic material previously identified by—and integrated into—the CRISPR-Cas system
The first stage, adaptation, leads to the insertion of new spacers in the CRISPR locus. In the second stage, expression, the system gets ready for action by expressing the cas genes and transcribing the CRISPR into a long precursor CRISPR RNA (pre-crRNA). The pre-crRNA is subsequently processed into mature crRNA by Cas proteins and accessory factors. In the third and last stage, interference, target nucleic acid is recognized and destroyed by the combined action of crRNA and Cas proteins
A.Price et al., (2016): CRISPR-Cas systems operate as adaptive immune defenses to target and degrade nucleic acids derived from bacteriophages and other foreign genetic elements. 12
1. Dana K Howe Muller's Ratchet and compensatory mutation in Caenorhabditis briggsae mitochondrial genome evolution 2008
2. Eugene V Koonin: Inevitability of Genetic Parasites 2016 Sep 26
3. Eugene V. Koonin: Inevitability of the emergence and persistence of genetic parasites caused by evolutionary instability of parasite-free states 04 December 2017
4. Gregory P Fournier: Ancient horizontal gene transfer and the last common ancestors 22 April 2015
5. Aude Bernheim The pan-immune system of bacteria: antiviral defence as a community resource 06 November 2019
6. Felix Broecker: Evolution of Immune Systems From Viruses and Transposable Elements 29 January 2019
7. Eugene V. Koonin: Evolution of adaptive immunity from transposable elements combined with innate immune systems December 2014
8. Eugene V. Koonin: The LUCA and its complex virome 14 July 2020
9. Luciano Marraffini: (Ph)ighting phages – how bacteria resist their parasites 2020 Feb 13
10. Simon J Labrie: Bacteriophage resistance mechanisms 2010 Mar 29.
11. Anna Lopatina: Abortive Infection: Bacterial Suicide as an Antiviral Immune Strategy 2020 Sep 29
12. Aryn A. Price et al.,: Harnessing the Prokaryotic Adaptive Immune System as a Eukaryotic Antiviral Defense 2016 Feb 3
13. Devashish Rath: The CRISPR-Cas immune system: Biology, mechanisms and applications October 2015
14. Dipali G Sashital: The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation Apr 25, 2019
15. SIMON A. JACKSON: CRISPR-Cas: Adapting to change 7 Apr 2017
16. M. P. Terns et al. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furious 2015 Jun; 21
17. Carl Zimmer Breakthrough DNA Editor Born of Bacteria February 6, 2015
18. Jordana Cepelewicz: Biodiversity Alters Strategies of Bacterial Evolution January 6, 2020
19. Tina Y.Liu: Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation 16 October 2020
20. Giedrius Gasiunas: Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria September 4, 2012
21. Samuel H. Sternberg et al. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System 2015 Nov 5
22. Eugene V Koonin: Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPRCas systems 2011 Jul 14
23. Eugene V Koonin: Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants 19 December 2019
24. Pascale Cossart: THE NEW Microbiology From Microbiomes to CRISPR 2016
25. Luciano A. Marraffini: Molecular mechanisms of CRISPR–Cas spacer acquisition 31 August 2018
26. Udi Qimron: Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli 2012 Feb 8
27. John van der Oost et al.: Unravelling the structural and mechanistic basis of CRISPR–Cas systems 09 June 2014
https://www.the-scientist.com/news-opinion/prokaryotes-are-capable-of-learning-to-recognize-phages-70378
https://reasonandscience.catsboard.com/t3243-origin-of-crispr-cas-molecular-complexes-of-prokaryotes
1. All entrance identity check systems based on data collection and storage mechanisms are designed.
2. CRISPR-Cas is an immune system based on data storage and identity check systems.
3. Therefore, it was designed.
Casey Luskin: Researchers Suggest Molecular Machine Is Irreducibly Complex August 8, 2014,
https://evolutionnews.org/2014/08/researchers_sug/
Structure of molecular machine that targets viral DNA for destruction determined
"The structure of this biological machine is conceptually similar to an engineer's blueprint, and it explains how each of the parts in this complex assemble into a functional complex that efficiently identifies viral DNA when it enters the cell," Wiedenheft said. "This surveillance machine consists of 12 different parts and each part of the machine has a distinct job. If we're missing one part of the machine, it doesn't work."
https://www.sciencedaily.com/releases/2014/08/140807154143.htm
R.N. JACKSON: Crystal structure of the CRISPR RNA–guided surveillance complex from Escherichia coli 7 Aug 2014
https://www.science.org/doi/10.1126/science.1256328
Programmable Memory in Prokaryotes April 12, 2017
https://evolutionnews.org/2017/04/programmable-memory-in-prokaryotes/
Luciano A. Marraffini (2018): Many bacteria and archaea have the unique ability to heritably alter their genomes by incorporating small fragments of foreign DNA, called spacers, into CRISPR loci. Once transcribed and processed into individual CRISPR RNAs, spacer sequences guide Cas effector nucleases to destroy complementary, invading nucleic acids. Collectively, these two processes are known as the CRISPR–Cas immune response. 25
Udi Qimron: CRISPR and their associated proteins comprise a significant prokaryotic defense system against viruses and horizontally transferred nucleic acids 26
https://www.youtube.com/watch?v=qc6xgb4VXl0
CRISPR is actually a sequence of information that is present in the bacterial genome and it is regularly interspersed and repetitive in nature that's why it's called palindromic repeats and there is a protein that is associated with CRISPR locus which is known as CRISPR associated protein line which works like a molecular scissor. CRISPR can be understood as a defense mechanism against viruses that are used against phage viruses so the phage viruses are the key enemies of e.coli which infect them and kill them so the virus has developed a defense mechanism that is embedded in their genome and which is in terms of a DNA sequence CRISPR locus and a cas gene which is producing a protein cas9. The sequence information of the CRISPR locus is strikingly similar to the sequence information present in the phage genome scientists scratch their heads why this sequence information is identical and similar what is the consequence of that or what is the cause of that? The phage infected the bacteria and the fed genetic material is inside the bacteria and tries to get integrated into the bacterial genome these sequences remain incorporated in the CRISPR locus now once the sequence gets incorporated into the CRISPR locus they become a part of the bacterial genome. Let's say there is a second infection by the same patch now this CRISPR locus would create specific RNA which is targeting the phage genome and the cas gene product which is cas9 would get associated with that RNA which is produced by the CRISPR locus called guide RNA and another RNA called tracer RNA and ultimately this cas9 along with this tracer and guide RNA cleaves the fudge genome now once the phage genome is cleaved using this CRISPR cas9 system the phage DNA would be degraded and that is how the bacteria protects itself from phage viruses and that's how it works it's a bacterial adaptive immune system so precise and beautiful.
Pascale Cossart (2016): In nature, bacteria need to defend themselves constantly, particularly against bacteriophages (or phages), the viruses that specifically attack bacteria. A phage generally attaches itself to a bacterium, injects its DNA into it, and subverts the bacterium’s mechanisms of replication, transcription, and translation in order to replicate itself. The phage DNA reproduces its own DNA, transcribes it into RNA, and produces phage proteins that accumulate to generate new phages and eventually cause the bacterial cell to explode (or lyse), releasing hundreds of new bacteriophages. Phages continually infect bacteria everywhere—in soil, in water, and even in our own intestinal microbiota. Bacteriophage families are numerous and vary widely in their form, size, composition, and the bacteria they target. To begin their attack, bacteriophages need a site of attachment, a particular component on the surface of a bacterium. This site of attachment is specific for each virus and the bacteria that it can infect. Bacteria have an immune system called CRISPR. CRISPR regions in the chromosomes allow bacteria to recognize predators, particularly previously encountered phages, and to destroy them. CRISPR regions protect and essentially “vaccinate” bacteria against bacteriophages. In fact, it has been shown that bacteria can be artificially vaccinated! When a population of bacteria is inoculated with a phage, a small number survive and are able to integrate a fragment of the phage DNA into their genome, in the region called the CRISPR locus. This allows the bacteria, if the phage ever attacks again, to recognize the phage DNA and degrade it. This ingenious phenomenon, known as interference, occurs due to the structure of the CRISPR region and to cas genes (CRISPR-associated genes) located near this region. The CRISPR locus is a region of the chromosome composed of repeated sequences of around 50 nucleotides, interspersed with sequences known as spacers that are similar to those of bacteriophages. Some bacteria have several CRISPR loci with different sequence repetitions. Around 40% of bacteria have one or more CRISPRs, whereas others have none. CRISPR loci can be quite long, sometimes with more than 100 repetitions and spacers. CRISPRs have two functions: acquisition and interference. Acquisition, also called adaptation, is the process of acquiring fragments of DNA from a phage, and interference is the immunization process by Cas proteins encoded by cas genes (Fig. below).

(Top) The three steps involved in CRISPR function.
(1) Integration of a piece of DNA from a phage into the CRISPR locus (acquisition);
(2) the expression of Cas proteins and of pre-crRNA, which is then split into small crRNAs;
(3) the interference that takes place when the DNA injected by a phage into a bacterium meets a crRNA, a hybrid form that is then degraded, consequently preventing infection.
(Bottom) Schematic drawing of genome modification (gene editing) by an sgRNA (small guided RNA) made of crRNA and tracrRNA and the endonuclease Cas9.
Bacteria have numerous proteins with various complementary and synergistic functions in the process of adaptation and interference. They permit the addition of DNA fragments into the CRISPR locus, but their main purpose is to react to invading phages. The CRISPR locus is transcribed into a long CRISPR RNA, which is then split into small RNAs called crRNAs, each containing a spacer and a part of the repeated sequence. When a phage injects its DNA into the bacterium, the crRNA recognizes and binds to it. An enzyme then recognizes the hybrid and cleaves the phage DNA at the point where the crRNA has paired. Replication of the phage DNA is inactivated, and the infection is stopped. Genome editing or modification is the identification of the proteins involved in the cleavage of the hybrid DNA. This process is performed by a complex of proteins containing the protein Cas1 and sometimes by a single protein called Cas9. Cas9 is unique in that it can attach itself to a DNA strand and, due to the two distinct domains of its structure, cut this DNA on each of its two strands. This protein is the basis of the CRISPR/Cas9 technology, which enables a variety of genome modifications and mutations in mammals, plants, insects, and fish in addition to bacteria. This system works due to the Cas9 protein and also a guide RNA hybrid that is made from one RNA similar to the region to be mutated and a second RNA called tracrRNA, or trans-activating crRNA. tracrRNA was discovered next to the CRISPR locus in Streptococcus pyogenes and was shown to be homologous to the repeated regions of the locus, enabling it to guide the Cas9 protein and the crRNA toward the target. In summary, by expressing the Cas9 protein with a composite RNA made up of an identical sequence to the target region, a tracrRNA, and a complementary fragment to the tracrRNA, one can now introduce a mutation or deletion into a target genome of any origin. After the 2012 publication in Science of the elegant studies by the teams led by Emmanuelle Charpentier and Jennifer Doudna, the CRISPR method was so intriguing that it provoked an avalanche of research and publications demonstrating that this technique could be used in many cases and with many variations. 24
The CRISPR–Cas system
Tina Y.Liu (2020): CRISPR-Cas systems stand out as the only known RNA programmed pathways for detecting and destroying bacteriophages and plasmids. . Class 1 CRISPR-Cas systems, the most widespread and diverse of these adaptive immune systems, use an RNA-guided multi-protein complex to find foreign nucleic acids and trigger their destruction. These multisubunit complexes target and cleave DNA and RNA, and regulatory molecules control their activities. CRISPR-Cas loci constitute the only known adaptive immune system in bacteria and archaea. They typically include an array of repeat sequences (CRISPRs) with intervening “spacers” matching sequences of DNA or RNA from viruses or other mobile genetic elements, and a set of genes encoding CRISPR-associated (Cas) proteins.
Transcription across the CRISPR array produces a precursor crRNA (pre-crRNA) that is processed by nucleases into small, non-coding CRISPR RNAs (crRNAs). Each crRNA molecule assembles with one or more Cas proteins into an effector complex that binds crRNA-complementary regions in foreign DNA or RNA. The effector complex then triggers degradation of the targeted DNA or RNA using either an intrinsic nuclease activity or a separate nuclease in trans.
Giedrius Gasiunas (2012):The silencing of invading nucleic acids is executed by ribonucleoprotein complexes preloaded with small, interfering CRISPR RNAs (crRNAs) that act as guides for targeting and degradation of foreign nucleic acid. The Cas9–crRNA complex of the Streptococcus thermophilus CRISPR3/Cas system introduces a double-strand break at a specific site in DNA containing a sequence complementary to crRNA. DNA cleavage is executed by Cas9, which uses two distinct active sites, RuvC and HNH, to generate site-specific nicks on opposite DNA strands. Results demonstrate that the Cas9–crRNA complex functions as an RNA-guided endonuclease with RNA-directed target sequence recognition and protein-mediated DNA cleavage. 20
J.Cepelewicz (2020): CRISPR acts like an adaptive immune system; it enables bacteria that have been exposed to a virus to pass on a genetic “memory” of that infection to their descendants, which can then mount better defenses against a repeat infection. It’s a system that works so well that an estimated half of all bacterial species use CRISPR. Researchers have uncovered dozens of other systems that bacteria use to rebuff phage invasions. But in laboratory studies, bacteria primarily develop what’s known as surface-based phage resistance. Mutations change receptor molecules on the surface of the bacterial cell, so that the phage can no longer recognize and invade it.
The strategy is akin to shutting a door and throwing away the key: It offers the bacteria complete safety from infection by the virus. But that protection comes at a significant price, because it also disrupts whatever nutrient uptake, waste disposal, communication task or other cellular function the receptor would have been providing — taking a constant toll on a cell’s fitness.
In contrast, CRISPR only drags on a cell’s resources when it’s active, during a viral infection. Even so, CRISPR represents a riskier gambit: It doesn’t start to work until phages have already entered the cell, meaning that there’s a chance the viruses could overcome it. And CRISPR doesn’t just attack viral DNA; it can also prevent bacteria from taking up beneficial genes from other microbes, like those that confer antibiotic resistance. What factors affect the trade-offs in costs and fitness? For the past six years, Edze Westra, an evolutionary ecologist at the University of Exeter in England, has led a team pursuing the answer to that question. In 2015, they discovered that nutrient availability and phage density affected whether Pseudomonas bacteria relied on surface-based or CRISPR-based resistance. In environments poor in resources, receptor modifications were more burdensome, so CRISPR became a better bargain. When resources were plentiful, bacteria grew more densely and phage epidemics became more frequent. Bacteria then faced greater selective pressure to close themselves off from infection entirely, and so they shut down receptors to gain surface-based resistance. This explained why surface-based resistance was so common in laboratory cultures. Growing in a test tube rich in nutrients, “these bacteria are on a holiday,” Westra said. “They are having a terrific time.”
Still, these rules weren’t cut and dried. Plenty of bacteria in natural high-nutrient environments use CRISPR, and plenty of bacteria in natural low-nutrient environments don’t. “It’s all over the place,” Westra said. “That told us that we were probably still missing something.”
How Biodiversity Reshapes the Battle
Then one of Westra’s graduate students, Ellinor Opsal, proposed another potential factor: the diversity of the biological communities in which bacteria live. This factor is harder to study, but scientists had previously observed that it could affect phage immunity in bacteria. For example, in 2005, James Bull, a biologist at the University of Texas, Austin, and William Harcombe, his graduate student at the time (now at the University of Minnesota), found that E. coli bacteria didn’t evolve immunity to a phage when a second bacterial species was present. Similarly, Britt Koskella, an evolutionary biologist at the University of California, Berkeley, and one of her graduate students, Catherine Hernandez, reported last year that phage resistance failed to arise in Pseudomonas bacteria living on their actual host (a plant), though they always gained immunity in a test tube. Could the diversity of the surroundings influence not just whether or not resistance to phages evolved, but the nature of that resistance?
To find out, Westra’s team performed a new set of experiments: Instead of altering the nutrient conditions for Pseudomonas bacteria growing with phages, they added three other bacterial species — species that competed against Pseudomonas for resources but weren’t targeted by the phage. Left to themselves, Pseudomonas would normally develop surface-based mutations. But in the company of rivals, they were far more likely to turn to CRISPR. Further investigation showed that the more complex community dynamics had shifted the fitness costs: The bacteria could no longer afford to inactivate receptors because they not only had to survive the phage, but also had to outcompete the bacteria around them. These results from Westra’s group dovetail with earlier findings that phages can produce greater diversity in bacterial communities. “Now, that diversity is actually feeding back to the phage side of things” by affecting phage resistance, Koskella said. “It’s neat to see that coming full circle.” By understanding that kind of feedback loop, she added, “we can start to ask more general questions about the impacts that phages have in a community context.”
For one, the bacteria’s shift toward a CRISPR-based phage response had another, broader effect. When Westra’s group grew Pseudomonas in moth larvae hosts, they found that the bacteria with surface-based resistance were less virulent, killing the larvae much more slowly than the bacteria with active CRISPR systems did. 18
Discovering CRISPR
S.H. Sternberg (2015): The CRISPR locus was first identified in Escherichia coli as an unusual series of 29-bp repeats separated by 32-bp spacer sequences (Ishino et al., 1987) 21 Carl Zimmer tells us the story (2015): The scientists who discovered CRISPR had no way of knowing that they had discovered something so revolutionary. They didn’t even understand what they had found. In 1987, Yoshizumi Ishino and colleagues at Osaka University in Japan published the sequence of a gene called iap belonging to the gut microbe E. coli. To better understand how the gene worked, the scientists also sequenced some of the DNA surrounding it. They hoped to find spots where proteins landed, turning iap on and off. But instead of a switch, the scientists found something incomprehensible. Near the iap gene lay five identical segments of DNA. DNA is made up of building blocks called bases, and the five segments were each composed of the same 29 bases. These repeat sequences were separated from each other by 32-base blocks of DNA, called spacers. Unlike the repeat sequences, each of the spacers had a unique sequence.
This peculiar genetic sandwich didn’t look like anything biologists had found before. When the Japanese researchers published their results, they could only shrug. “The biological significance of these sequences is not known,” they wrote. It was hard to know at the time if the sequences were unique to E. coli, because microbiologists only had crude techniques for deciphering DNA. But in the 1990s, technological advances allowed them to speed up their sequencing. By the end of the decade, microbiologists could scoop up seawater or soil and quickly sequence much of the DNA in the sample. This technique — called metagenomics — revealed those strange genetic sandwiches in a staggering number of species of microbes. They became so common that scientists needed a name to talk about them, even if they still didn’t know what the sequences were for. In 2002, Ruud Jansen of Utrecht University in the Netherlands and colleagues dubbed these sandwiches “clustered regularly interspaced short palindromic repeats” — CRISPR for short.
Jansen’s team noticed something else about CRISPR sequences: They were always accompanied by a collection of genes nearby. They called these genes Cas genes, for CRISPR-associated genes. The genes encoded enzymes that could cut DNA, but no one could say why they did so, or why they always sat next to the CRISPR sequence. Three years later, three teams of scientists independently noticed something odd about CRISPR spacers. They looked a lot like the DNA of viruses. “And then the whole thing clicked,” said Eugene Koonin. At the time, Koonin, an evolutionary biologist at the National Center for Biotechnology Information in Bethesda, Md., had been puzzling over CRISPR and Cas genes for a few years. As soon as he learned of the discovery of bits of virus DNA in CRISPR spacers, he realized that microbes were using CRISPR as a weapon against viruses.
Koonin knew that microbes are not passive victims of virus attacks. They have several lines of defense. Koonin thought that CRISPR and Cas enzymes provide one more. In Koonin’s hypothesis, bacteria use Cas enzymes to grab fragments of viral DNA. They then insert the virus fragments into their own CRISPR sequences. Later, when another virus comes along, the bacteria can use the CRISPR sequence as a cheat sheet to recognize the invader.
Scientists didn’t know enough about the function of CRISPR and Cas enzymes for Koonin to make a detailed hypothesis. But his thinking was provocative enough for a microbiologist named Rodolphe Barrangou to test it. To Barrangou, Koonin’s idea was not just fascinating, but potentially a huge deal for his employer at the time, the yogurt maker Danisco. Danisco depended on bacteria to convert milk into yogurt, and sometimes entire cultures would be lost to outbreaks of bacteria-killing viruses. Now Koonin was suggesting that bacteria could use CRISPR as a weapon against these enemies.
To test Koonin’s hypothesis, Barrangou and his colleagues infected the milk-fermenting microbe Streptococcus thermophilus with two strains of viruses. The viruses killed many of the bacteria, but some survived. When those resistant bacteria multiplied, their descendants turned out to be resistant too. Some genetic change had occurred. Barrangou and his colleagues found that the bacteria had stuffed DNA fragments from the two viruses into their spacers. When the scientists chopped out the new spacers, the bacteria lost their resistance. Barrangou, now an associate professor at North Carolina State University, said that this discovery led many manufacturers to select for customized CRISPR sequences in their cultures, so that the bacteria could withstand virus outbreaks. “If you’ve eaten yogurt or cheese, chances are you’ve eaten CRISPR-ized cells,” he said.
In 2007, Blake Wiedenheft joined Doudna’s lab as a postdoctoral researcher, eager to study the structure of Cas enzymes to understand how they worked. Doudna agreed to the plan — not because she thought CRISPR had any practical value, but just because she thought the chemistry might be cool. “You’re not trying to get to a particular goal, except understanding,” she said. As Wiedenheft, Doudna and their colleagues figured out the structure of Cas enzymes, they began to see how the molecules worked together as a system. When a virus invades a microbe, the host cell grabs a little of the virus’s genetic material, cuts open its own DNA, and inserts the piece of virus DNA into a spacer. As the CRISPR region fills with virus DNA, it becomes a molecular most-wanted gallery, representing the enemies the microbe has encountered. The microbe can then use this viral DNA to turn Cas enzymes into precision-guided weapons. The microbe copies the genetic material in each spacer into an RNA molecule. Cas enzymes then take up one of the RNA molecules and cradle it. Together, the viral RNA and the Cas enzymes drift through the cell. If they encounter genetic material from a virus that matches the CRISPR RNA, the RNA latches on tightly. The Cas enzymes then chop the DNA in two, preventing the virus from replicating.
CRISPR, microbiologists realized, is also an adaptive immune system. It lets microbes learn the signatures of new viruses and remember them. And while we need a complex network of different cell types and signals to learn to recognize pathogens, a single-celled microbe has all the equipment necessary to learn the same lesson on its own. But how did microbes develop these abilities? Ever since microbiologists began discovering CRISPR-Cas systems in different species, Koonin and his colleagues have been reconstructing the systems’ evolution. CRISPR-Cas systems use a huge number of different enzymes, but all of them have one enzyme in common, called Cas1. The job of this universal enzyme is to grab incoming virus DNA and insert it in CRISPR spacers. Recently, Koonin and his colleagues discovered what may be the origin of Cas1 enzymes.
Along with their own genes, microbes carry stretches of DNA called mobile elements that act like parasites. The mobile elements contain genes for enzymes that exist solely to make new copies of their own DNA, cut open their host’s genome, and insert the new copy. Sometimes mobile elements can jump from one host to another, either by hitching a ride with a virus or by other means, and spread through their new host’s genome.
Koonin and his colleagues discovered that one group of mobile elements, called casposons, makes enzymes that are pretty much identical to Cas1. In a new paper in Nature Reviews Genetics, Koonin and Mart Krupovic of the Pasteur Institute in Paris argue that the CRISPR-Cas system got its start when mutations transformed casposons from enemies into friends. Their DNA-cutting enzymes became domesticated, taking on a new function: to store captured virus DNA as part of an immune defense. While CRISPR may have had a single origin, it has blossomed into a tremendous diversity of molecules. Koonin is convinced that viruses are responsible for this. Once they faced CRISPR’s powerful, precise defense, the viruses evolved evasions. Their genes changed sequence so that CRISPR couldn’t latch onto them easily. And the viruses also evolved molecules that could block the Cas enzymes. The microbes responded by evolving in their turn. They acquired new strategies for using CRISPR that the viruses couldn’t fight. Over many thousands of years, in other words, evolution behaved like a natural laboratory, coming up with new recipes for altering DNA. 17
Diversity, ecology, and evolution of the CRISPR-Cas systems
Devashish Rath (2015): The length and sequence of repeats and the length of spacers are well conserved within a CRISPR locus, but may vary between CRISPRs in the same or different genomes. Repeat sequences are in the range of 21 bp to 48 bp, and spacers are between 26 bp and 72 bp. The observed variation is perhaps not surprising given how widespread the system is. The number of spacers within a CRISPR locus varies widely; from a few to several hundred. Genomes can have single or multiple CRISPR loci and in some species, these loci can make up a significant part of the chromosome. Not all CRISPR loci have adjacent cas genes and instead rely on trans-encoded factors. (a trans-acting factor is usually a regulatory protein that binds to DNA). Another feature associated with CRISPR loci is the presence of a conserved sequence, called leader, located upstream of the CRISPR with respect to the direction of transcription. The Cas proteins are a highly diverse group. Many are predicted or identified to interact with nucleic acids; e.g. as nucleases, helicases and RNA-binding proteins. The Cas1 and Cas2 proteins are involved in adaptation and are virtually universal for CRISPR-Cas systems. Other Cas proteins are only associated with certain types of CRISPR-Cas systems. The diversity of Cas proteins, presence of multiple CRISPR loci, and frequent horizontal transfer of CRISPR-Cas systems make classification a complex task. The most adopted classification identifies Type I, II and III CRISPR-Cas systems, with each having several subgroups. Different types of CRISPR-Cas systems can co-exist in a single organism. Recently, a Type IV system was proposed, which contains several Cascade genes but no CRISPR, cas1 or cas2. Type IV complex would be guided by protein-DNA interaction, not by crRNA, and constitutes an innate immune system preset to attack certain sequences. The Type I systems are defined by the presence of the signature protein Cas3, a protein with both helicase and DNase domains responsible for degrading the target. Currently, six subtypes of the Type I system are identified (Type I-A through Type I-F) that have a variable number of cas genes. Apart from cas1, cas2 and cas3, all Type I systems encode a Cascade-like complex. Cascade binds crRNA and locates the target, and most variants are also responsible for processing the crRNA. Cascade also enhances spacer acquisition in some cases. In the Type I-A system, Cas3 is a part of the Cascade complex. The Type II CRISPR-Cas systems encode Cas1 and Cas2, the Cas9 signature protein, and sometimes a fourth protein (Csn2 or Cas4). Cas9 assists in adaptation participate in crRNA processing and cleave the target DNA assisted by crRNA and an additional RNA called tracrRNA. Type II systems have been divided into subtypes II-A and II-B but recently a third, II-C, has been suggested. The csn2 and cas4 genes, both encoding proteins involved in adaptation, are present in Type II-A and Type II-B, respectively, while Type II-C lacks a fourth gene. The Type III CRISPR-Cas systems contain the signature protein Cas10 with unclear function. Most Cas proteins are destined for the Csm (in Type III-A) or Cmr (in Type III-B) complexes, which are similar to Cascade. Interestingly, while all Type I and II systems are known to target DNA, Type III systems target DNA and/or RNA. So far, the Type II systems have been exclusively found in bacteria while the Type I and Type III systems occur both in bacteria and archaea. A large number of genomes with detected CRISPRs could be used as an argument for its importance as a defense mechanism. However, the CRISPR-Cas systems are probably mobile genetic elements that frequently transfer horizontally, which also contributes to their high prevalence. Other findings indicate that phages can still replicate in populations with one but not two spacers targeting them.
Eugene V. Koonin (2019): The number and diversity of known CRISPR–Cas systems have substantially increased in recent years. The new classification includes 2 classes, 6 types and 33 subtypes compared with 5 types and 16 subtypes in 2015. At the adaptation stage, a distinct complex of Cas proteins binds to a target DNA, often after recognizing a distinct, short motif known as a protospacer-adjacent motif (PAM), and cleaves out a portion of the target DNA, the protospacer. After duplication of the repeat at the 5ʹ end of the CRISPR array, the adaptation complex inserts the protospacer DNA into the array, so that it becomes a spacer. Some CRISPR–Cas systems employ an alternative mechanism of adaptation — namely, spacer acquisition from RNA, via reverse transcription by a reverse transcriptase encoded at the CRISPR–cas locus. At the expression stage, the CRISPR array is typically transcribed as a single transcript — the pre-CRISPR RNA (pre-crRNA) — that is processed into mature CRISPR RNAs (crRNAs), each containing the spacer sequence and parts of the flanking repeats. In different CRISPR–Cas variants, the pre-crRNA processing is mediated by a distinct subunit of a multiprotein Cas complex, by a single, multidomain Cas protein, or by non-Cas host RNases. At the interference stage, the crRNA, which typically remains bound to the processing complex (protein), serves as a guide to recognize the protospacer (or a closely similar sequence) in the invading genome of a virus or plasmid, which is then cleaved and inactivated by a Cas nuclease (or nucleases) that either is part of the effector or is recruited at the interference stage. The above summary is a brief, oversimplified description of the CRISPR–Cas functionality that inevitably omits many details. These can be found in recent reviews on different aspects of CRISPR–Cas biology. Similar to other biological defense mechanisms, archaeal and bacterial CRISPR–Cas systems show a remarkable diversity of Cas protein sequences, gene compositions, and architectures of the genomic loci. Our knowledge of this diversity is continuously expanding through the screening of ever-growing genomic and metagenomic databases. To keep pace with such expansion, a robust classification of CRISPR–Cas systems is essential for the progress of CRISPR research, but this presents formidable challenges, owing to the lack of universal markers and the fast evolution of the CRISPR–cas loci. Therefore, the two previous CRISPR–Cas classifications, published in Nature Reviews Microbiology in 2011 and 2015, employed a multipronged approach that combined comparisons of the gene compositions of CRISPR–Cas systems and their loci architectures with sequence similarity-based clustering and phylogenetic analysis of conserved Cas proteins, such as Cas1. The 2015 classification included 5 types and 16 subtypes, as well as introducing the major division of CRISPR–Cas systems into two classes that radically differ with respect to the architectures of their effector modules involved in crRNA processing and interference. The class 1 systems have effector modules composed of multiple Cas proteins, some of which form crRNA-binding complexes (such as the Cascade complex in type I systems) that, with contributions from additional Cas proteins, mediate pre-crRNA processing and interference. By contrast, class 2 systems encompass a single, multidomain crRNA-binding protein (such as Cas9 in type II systems) that combines all activities required for interference and, in some variants, also those involved in pre-crRNA processing (Box 1).

Class 1 CrisPr–Cas systems have effector modules composed of multiple Cas proteins that form a crrNa-binding complex and function together in binding and processing of the target. Class 2 systems have a single, multidomain. crrNa-binding protein that is functionally analogous to the entire effector complex of class 1. Part a of the figure illustrates the generic organizations of the class 1 and class 2 CrisPr–Cas loci. Part b of the figure shows the functional modules of CrisPr–Cas systems. the scheme shows the typical relationships between the genetic, structural and functional organizations of the six types of CrisPr–Cas systems. Protein names follow the current nomenclature. an asterisk indicates the putative small subunit that might be fused to the large subunit in several type i subtypes. the pound symbols (#) indicate that other unknown sensor, effector and ring nuclease protein families could be involved in the same signaling pathway. Dispensable (and/or missing, in some subtypes and variants) components are indicated by dashed outlines. Cas6 is shown with a thin solid outline for type i because it is dispensable in some, but not most, systems and with a dashed line for type iii because most of these systems apparently use the Cas6 protein provided in trans by other CrisPr–cas loci. the three colours for Cas9, Cas10, Cas12 and Cas13 reflect the fact that these proteins contribute to different stages of the CrisPr–Cas response. the CrisPr-associated rossmann fold (CarF) and higher eukaryotes and prokaryotes nucleotide-binding (HePN) domain proteins are the most common sensors and effectors, respectively, in the type III ancillary modules, but several alternative sensors and effectors have been identified, as well43. ring nucleases are a distinct variety of CarF domain proteins that cleave cyclic oligoa produced by Cas10 and thus control the indiscriminate rNase activity of the HePN domain of Csx1. Ls, large subunit; ss, small subunit; tracrrNa, transactivating CrisPr rNa.
CRISPR–Cas classification
No genes are shared by all CRISPR–Cas systems, ruling out the possibility of a straightforward, comprehensive phylogenetic classification analogous to that employed for cellular life forms.
Class 1 and its derivatives.
The classification of class 1 CRISPR–Cas systems, which include types I, III and IV, has remained relatively stable compared with the 2015 version (Fig. below).

Updated classification of class 1 criSPr–cas systems.
The figure schematically shows representative (typical) CRISPR–cas loci of each class 1 subtype and of selected distinct variants, with the dendrogram on the left showing the likely evolutionary relationships between the types and subtypes. The column on the right indicates the organism and the corresponding gene range. Homologous genes are colour-coded and identified by a family name. The gene names follow the previous classification18. Where both a systematic name and a legacy name are commonly used, the legacy name is given under the systematic name. The small subunit is encoded by csm2, cmr5, cse2, csa5 and several additional families of homologous genes that are collectively denoted cas11. The adaptation module genes cas1 and cas2 are dispensable in subtypes III-A and III-E (dashed lines). Gene regions coloured cream represent the HD nuclease domain; the HD domain in Cas10 is distinct from that in Cas3 and Cas3ʹʹ. Functionally uncharacterized genes are shown in grey. The tan shading shows the effector module. The grey shading of different hues shows the two levels of classification: subtypes and variants. Most of the subtype III-B, III-C, III-E and III-F loci, as well as IV-B and IV-C loci, lack CRISPR arrays and are shown accordingly, although for each of the type III subtypes exceptions have been detected. CHAT, protease domain of the caspase family; RT, reverse transcriptase; TPR, tetratricopeptide repeat.

Updated classification of class 2 criSPr–cas systems.
The figure schematically shows representative (typical) CRISPR–cas loci for each class 2 subtype and for selected distinct variants, with the dendrogram on the left showing the likely evolutionary relationships between the types and subtypes. The column on the right indicates the organism and the corresponding gene range. Homologous genes are colour coded and are identified by a family name following the previous classification18. Where both a systematic name and a legacy name are commonly used, the legacy name is given under the systematic name. The grey shading of different hues shows the two levels of classification: subtypes and variants. The adaptation module genes cas1 and cas2 are present in only a subset of the subtype V-D, VI-A and VI-D loci and are accordingly shown by dashed lines. The WYL-domain-encoding genes and csx27 genes are also dispensable and shown by dashed lines. Additional genes encoding components of the interference module, such as transactivating CRISPR RNA (tracrRNA), are shown. The domains of the effector proteins are colour-coded: RuvC-like nuclease, green; HNH nuclease, yellow; higher eukaryotes and prokaryotes nucleotide-binding (HEPN) RNase, purple; transmembrane domains, blue.

Outline of a complete scenario for the origins and evolution of criSPr–cas systems.
The figure depicts a hypothetical scenario of the origin of CRISPR–Cas systems from an ancestral signalling system (possibly an abortive infection defence system (Abi)). This putative ancestral Abi module shares a cyclic oligoA polymerase Palm domain (RNA recognition motif (RRM) fold) with Cas10 and is proposed to function analogously to type III CRISPR–Cas systems. Specifically, cyclic oligoA molecules that are synthesized in response to virus infection bind to the CRISPR-associated Rossmann fold (CARF) domain of the second protein in this system, resulting in activation of the RNase activity of the higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domain, which induces dormancy through indiscriminate RNA cleavage. This putative ancestral Abi module would give rise to the type III-like CRISPR–Cas effector module via duplication of the RRM domain, with subsequent inactivation of one of the copies (the two RRM domains are denoted RRM1 and RRM2). The ancestral class 1 CRISPR–Cas system is inferred to have evolved through the merger of two modules: the adaptation module, including the CRISPR repeats, derived from a casposon, and the type III-like effector module, likely derived from the ancestral Abi system. The subsequent acquisition of the HD nuclease domain by the effector module provided for RNA-guided DNA cleavage. Inactivation of the oligoA polymerase domain in the effector complex, or possibly replacement of Cas10 by an unrelated protein and acquisition of the Cas3 helicase, led to the emergence of type I systems, which lack the cyclic oligoA-dependent signalling pathway and exclusively cleave double-stranded DNA. Class 2 systems of type II and different subtypes of type V appear to have evolved independently by the recruitment of distinct TnpB nucleases that are encoded by IS605-like transposable elements. Type VI likely originated from an RNA-cleaving, HEPN domain-containing abortive infection or toxin–antitoxin system. Some CRISPR–Cas systems, such as type IV and Tn7-linked systems I-F3 and V-K, were subsequently recruited by mobile genetic elements and lost their interference capacity along with the original defence function. The key evolutionary events are described to the right of the images. The typical CRISPR–cas operon organization is shown for each CRISPR–Cas subtype and for selected distinct variants. Homologous genes are colour-coded and identified by a family name following the previous classification18. The multiforking arrows denote events that have been inferred to have occurred on multiple, independent occasions during the evolution of CRISPR–Cas systems. GGDD, key catalytic motif of the cyclase or polymerase domain of Cas10 that is involved in the synthesis of cyclic oligoA signalling molecules; HRAMP, haloarchaeal repeat-associated mysterious proteins; TR, terminal repeats; tracrRNA, transactivating CRISPR RNA; TSD, target site duplication, the likely source of ancestral repeats. 23
Imagine a company had the task to install a security system in its headquarters, based on biometrics. Biometrics comes from the Greek words “bios” (life) and “metrikos” (measure). It involves the implementation of a system that uses the analysis of biological characteristics of people, and that analyzes human characteristics for identity verification or identification. In order to distinguish employees that are permitted to enter the building, and exclude to enter those that are not welcome, there has to be first data collection and storage of the information in a memory bank. Every time, when someone arrives at the building, it will go through the security check, and the provided data will be compared to the data in the memory bank. If there is a match, the person is permitted to enter, or not.
Analogously, cells are capable of doing almost the same, with a few differences. They have an ingenious security check system, based on enemy recognition, and based on that knowledge, creating a sophisticated data bank, that is employed to recognize future enemy invasions, and annihilate them.

Diversity of CRISPR–Cas systems.
The CRISPR-associated (Cas) proteins can be divided into distinct functional categories as shown. The three types of CRISPR–Cas systems are defined on the basis of a type-specific signature Cas protein (indicated by an asterisk) and are further subdivided into subtypes. The CRISPR ribonucleoprotein (crRNP) complexes of type I and type III systems contain multiple Cas subunits, whereas the type II system contains a single Cas9 protein. Boxes indicate components of the crRNP complexes for each system. The type III-B system is unique in that it targets RNA, rather than DNA, for degradation.
Understanding CRISPR-Cas9
Eugene V Koonin (2011): CRISPR-Cas systems have three distinct functional stages of their operation. During the first stage, adaptation, short pieces of DNA (characteristic length of approximately 30 bp) homologous to virus or plasmid sequences (known as proto-spacers) are integrated into the CRISPR loci. The short (3 or 4 nucleotides) proto-spacer adjacent motifs (PAMs) located immediately downstream of the proto-spacer appear to determine the selection of the protospacer followed by integration into a pre-existing CRISPR array. The second stage, expression and processing, involves transcription and cleavage of long primary transcript of a CRISPR locus (pre-crRNA) that is processed into short crRNAs. This step is catalyzed by endoribonucleases encoded by the cas genes that either operate as a subunit of a larger complex (e.g. Cascade, CRISPR-associated complex for antiviral defense in Escherichia coli) or as a stand-alone enzyme, e.g., Cas6 in the archaeon Pyrococcus furiosus. At the third stage, interference, the alien nucleic acid (DNA or RNA) is targeted by a ribonucleoprotein complex containing a crRNA guide and a set of Cas proteins, and cleaved within or in the vicinity of the PAM sequence. In several CRISPR-Cas systems, crRNA have been shown to be complementary to either strand of the phage or plasmid which is best compatible with DNA being the target. Direct demonstration of DNA being the target of the CRISPR-Cas machinery has come from experiments in Staphylococcus epidermidis. In this case, insertion of a self-splicing intron into the proto-spacer sequence of the target gene rendered the respective plasmid resistant to the CRISPR-mediated immunity. 22


A roadmap of CRISPR-Cas adaptation and defense.
In the example illustrated, a bacterial cell is infected by a bacteriophage. The first stage of CRISPR-Cas defense is CRISPR adaptation. This involves the incorporation of small fragments of DNA from the invader into the host CRISPR array. This forms a genetic “memory” of the infection. The memories are stored as spacers (colored squares) between repeat sequences (R), and new spacers are added at the leader-proximal (L) end of the array. The Cas1 and Cas2 proteins, encoded within the cas gene operon, form a Cas1-Cas2 complex (blue)—the “workhorse” of CRISPR adaptation. In this example, the Cas1-Cas2 complex catalyzes the addition of a spacer from the phage genome (purple) into the CRISPR array. The second stage of CRISPR-Cas defense involves transcription of the CRISPR array and subsequent processing of the precursor transcript to generate CRISPR RNAs (crRNAs). Each crRNA contains a single spacer unit that is typically flanked by parts of the adjoining repeat sequences (gray). Individual crRNAs assemble with Cas effector proteins (light green) to form crRNA-effector complexes. The crRNA-effector complexes catalyze the sequence-specific recognition and destruction of foreign DNA and/or RNA elements. This process is known as interference. 13
 [/size]
[/size]Overview of the CRISPR–Cas system.
Adaptive immunity by CRISPR–Cas systems is mediated by CRISPR RNAs (crRNAs) and Cas proteins, which form multicomponent CRISPR ribonucleoprotein (crRNP) complexes. The cas genes are colored according to function, as indicated by the four functional categories in coloured boxes:
spacer acquisition (yellow);
crRNA processing (pink);
crRNA assembly and surveillance (blue); and
target degradation (purple).
Involvement of non-Cas components (grey) is indicated, either when experimentally demonstrated (for example, RNase III processing in type II systems) or when anticipated (for example, the potential involvement of housekeeping repair and/or recombination enzymes). The first stage is known as acquisition, which occurs following the entry of an invading mobile genetic element (in this case, a viral genome). The invading DNA is fragmented and a new protospacer (green) is selected, processed and integrated as a new spacer at the leader end of the CRISPR array. During the second stage, which is known as expression, the CRISPR locus is transcribed and the pre-crRNA is processed into small crRNAs by CRISPR-associated (Cas6) and/or housekeeping ribonucleases (such as RNase III). The mature crRNAs and Cas proteins assemble to form a crRNP complex. During the final stage of interference, the crRNP scans invading DNA for a complementary nucleic acid target and on successful recognition, the target is eventually degraded by Cas nucleases. 27
S. H. Sternberg (2015): CRISPR-Cas immunity is conferred through integration of short DNA fragments into the CRISPR locus, and these spacer sequences record the history of past infections. The CRISPR locus is transcribed, and the resultant transcript is processed into shorter CRISPR-RNAs (crRNAs). CRISPR-Cas systems are classified as types I, II or III, which can be distinguished based on the presence of the signature Cas3, Cas9, or Cas10 genes, respectively. Type I are the most common, and much of our understanding of type I CRISPR-Cas systems comes from studies of E. coli Cascade (CRISPR-associated complex for antiviral defense), which is comprised of the five proteins Cse1, Cse2, Cas7, Cas5e, and Cas6e. These proteins assemble on a 61-nt crRNA, yielding a 405- kDa complex. The crRNA contains the 32-nt spacer sequence, which directs Cascade to sequences (protospacers) in foreign DNA, leading to formation of an R-loop intermediate. Cascade then recruits Cas3, which has an N-terminal histidine-aspartate (HD) nuclease domain and C-terminal superfamily 2 (SF2) helicase domain, to degrade the DNA. Cascade must discriminate between spacer sequences found in the bacterial chromosome and those found in foreign DNA. This discrimination is thought to be accomplished through recognition of a trinucleotide sequence motif called the protospacer-adjacent motif (PAM; 5′-A[A/T]G-3′ for E. coli Cascade), which is adjacent to the protospacer in foreign DNA, but absent in the CRISPR locus. Strict sequence requirements present a potential weakness because mutations in either the PAM or protospacer can allow foreign DNA to escape CRISPR-Cas immunity. However, bacteria can rapidly restore immunity using a positive-feedback loop to update the CRISPR locus. Priming requires Cascade with a crRNA bearing at least partial complementarity to the escape target, suggesting Cascade must be able to locate targets even when they bear mutations sufficient to escape immunity. Priming also requires Cas3 and the Cas1-Cas2 complex, which integrate new sequences into the CRISPR locus. The PAM-dependent pathway is highly efficient and allows Cascade to recruit Cas3 for strand-specific degradation of the target genome. The PAM-independent pathway is less efficient, but Cascade can still bind tightly to the DNA, ensuring that it can initiate the sequence of molecular events that precede primed spacer acquisition. Through this pathway, Cas3 recruitment becomes strictly dependent on Cas1-Cas2, and Cas1-Cas2 also attenuate Cas3 nuclease activity and enable Cas3 to rapidly translocate in either direction along the foreign DNA. These results establish Cas1-Cas2 as a trans-acting factor necessary for the recruitment and regulation of Cas3 at escape targets. Based on our findings, we propose a mechanistic framework describing how Cascade, Cas1, Cas2, and Cas3 work together to process and disable foreign genetic elements. 21
M. P. Terns et al. (2015): (CRISPR-Cas immune systems function to defend prokaryotes against potentially harmful mobile genetic elements including viruses and plasmids. The multiple CRISPR-Cas systems (Types I, II, and III) each target destruction of foreign nucleic acids via structurally and functionally diverse effector complexes (crRNPs). CRISPR-Cas effector complexes are comprised of CRISPR RNAs (crRNAs) that contain sequences homologous to the invading nucleic acids and Cas proteins specific to each immune system type. CRISPR-Cas systems confer prokaryotes with adaptive immunity against viruses, conjugative plasmids, and other potential genome invaders. A host CRISPR (clustered regularly interspaced short palindromic repeats) locus contains a leader region (typically 100–500 bp) followed by multiple copies of a repeat sequence (∼30–40 bp) separated by similarly sized, variable invader-derived sequences. Each crRNA contains a guide region comprised of invader-derived sequences that allow crRNA-Cas protein effector complexes to recognize and destroy invader nucleic acids. CRISPR-associated (Cas) proteins provide enzymatic machinery and structural components to carry out the distinct phases of the CRISPR-Cas pathway. Moreover, modules of Cas proteins (e.g., Csa, Cst, Cse, Csm, Cmr) comprise the distinct CRISPR-Cas immune systems: Type I (A-G), Type II (A-C), and Type III (A-B). 16
Dipali G Sashital (2019): Within this system, the CRISPR locus is programmed with ‘spacer’ sequences that are derived from foreign DNA and serve as a record of prior infection events 14 CRISPR cas9 in a bacteria acts as an adaptive immune response that is it remembers when a virus has infected the cell in the past and it keeps a little bit of viral DNA and stores it in a memory bank ( the spacers) and uses it so that if the same species of virus infects the cell again it will be able to compare the injected DNA to sequences in the data bank, recognize and respond to it quickly and effectively, and destroy it.
Devashish Rath (2015): The CRISPR-Cas mediated defense process can be divided into three stages.
1. Adaptation or spacer acquisition,where a short fragment of invading DNA is inserted into the CRISPR locus for future recognition of that invader;
2. crRNA biogenesis ( expression), which involves the biogenesis of guide RNA units (crRNA)
3. Target interference where these effector complexes vigilantly scan for and degrade invading genetic material previously identified by—and integrated into—the CRISPR-Cas system
The first stage, adaptation, leads to the insertion of new spacers in the CRISPR locus. In the second stage, expression, the system gets ready for action by expressing the cas genes and transcribing the CRISPR into a long precursor CRISPR RNA (pre-crRNA). The pre-crRNA is subsequently processed into mature crRNA by Cas proteins and accessory factors. In the third and last stage, interference, target nucleic acid is recognized and destroyed by the combined action of crRNA and Cas proteins
A.Price et al., (2016): CRISPR-Cas systems operate as adaptive immune defenses to target and degrade nucleic acids derived from bacteriophages and other foreign genetic elements. 12
1. Dana K Howe Muller's Ratchet and compensatory mutation in Caenorhabditis briggsae mitochondrial genome evolution 2008
2. Eugene V Koonin: Inevitability of Genetic Parasites 2016 Sep 26
3. Eugene V. Koonin: Inevitability of the emergence and persistence of genetic parasites caused by evolutionary instability of parasite-free states 04 December 2017
4. Gregory P Fournier: Ancient horizontal gene transfer and the last common ancestors 22 April 2015
5. Aude Bernheim The pan-immune system of bacteria: antiviral defence as a community resource 06 November 2019
6. Felix Broecker: Evolution of Immune Systems From Viruses and Transposable Elements 29 January 2019
7. Eugene V. Koonin: Evolution of adaptive immunity from transposable elements combined with innate immune systems December 2014
8. Eugene V. Koonin: The LUCA and its complex virome 14 July 2020
9. Luciano Marraffini: (Ph)ighting phages – how bacteria resist their parasites 2020 Feb 13
10. Simon J Labrie: Bacteriophage resistance mechanisms 2010 Mar 29.
11. Anna Lopatina: Abortive Infection: Bacterial Suicide as an Antiviral Immune Strategy 2020 Sep 29
12. Aryn A. Price et al.,: Harnessing the Prokaryotic Adaptive Immune System as a Eukaryotic Antiviral Defense 2016 Feb 3
13. Devashish Rath: The CRISPR-Cas immune system: Biology, mechanisms and applications October 2015
14. Dipali G Sashital: The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation Apr 25, 2019
15. SIMON A. JACKSON: CRISPR-Cas: Adapting to change 7 Apr 2017
16. M. P. Terns et al. Three CRISPR-Cas immune effector complexes coexist in Pyrococcus furious 2015 Jun; 21
17. Carl Zimmer Breakthrough DNA Editor Born of Bacteria February 6, 2015
18. Jordana Cepelewicz: Biodiversity Alters Strategies of Bacterial Evolution January 6, 2020
19. Tina Y.Liu: Chemistry of Class 1 CRISPR-Cas effectors: Binding, editing, and regulation 16 October 2020
20. Giedrius Gasiunas: Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria September 4, 2012
21. Samuel H. Sternberg et al. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System 2015 Nov 5
22. Eugene V Koonin: Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPRCas systems 2011 Jul 14
23. Eugene V Koonin: Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants 19 December 2019
24. Pascale Cossart: THE NEW Microbiology From Microbiomes to CRISPR 2016
25. Luciano A. Marraffini: Molecular mechanisms of CRISPR–Cas spacer acquisition 31 August 2018
26. Udi Qimron: Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli 2012 Feb 8
27. John van der Oost et al.: Unravelling the structural and mechanistic basis of CRISPR–Cas systems 09 June 2014
https://www.the-scientist.com/news-opinion/prokaryotes-are-capable-of-learning-to-recognize-phages-70378
Last edited by Otangelo on Wed Nov 02, 2022 6:13 pm; edited 43 times in total


















